Paramus App Store
Applications, Datasets, AI Models and Computational Models.
The Paramus App Store is a curated portfolio of HPC applications, AI models, datasets, and local LLMs for computational chemistry. All packages run on Paramus Chemistry OS, a Windows-based on-premise platform providing local compute power for demanding simulations. A subset of functionality is also available via Paramus Cloud, though the full App Store requires local hardware.
Featured Apps
*) We care deeply about legal compliance, and if you discover a license violation, please inform us at support@paramus.ai. We check on user registration, if the required licenses are present, otherwise you only get the free packages (which is still a lot). We are working on a central registration system with different vendors.
Browse Applications
Computing Applications (HPC) (33)
Computing Applications (HPC)
HPC enables scalable simulation, modeling, and analysis of chemical systems. In quantum chemistry (QC), HPC is crucial for performing accurate electronic structure calculations at high theory levels, enabling reliable predictions for molecular design and reactivity.

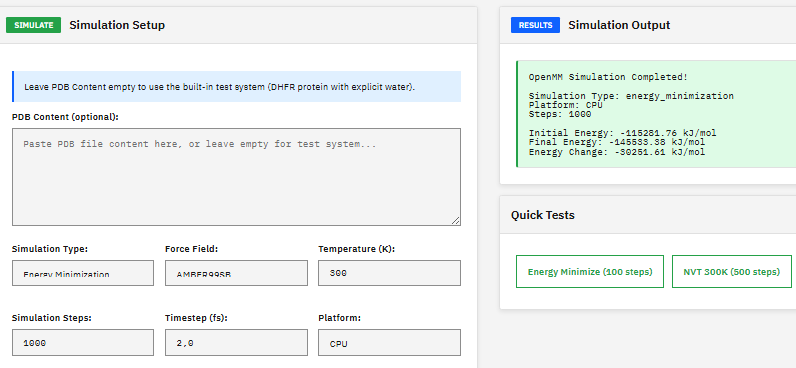
OpenMM
MIT (Free)
A molecular dynamics toolkit for simulating biomolecules, providing energy minimization, NVT/NPT ensemble sampling, and support for AMBER and CHARMM force fields with GPU-accelerated calculations via CUDA and OpenCL.
Features & Details
Key Features
- Force fields: AMBER, CHARMM, OPLS
- Ensembles: NVT, NPT, NVE
- GPU: CUDA, OpenCL acceleration
- Integrators: Langevin, Verlet, Brownian
Eastman, P. et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLOS Comput. Biol. 13, e1005659 (2017). DOI:10.1371/journal.pcbi.1005659
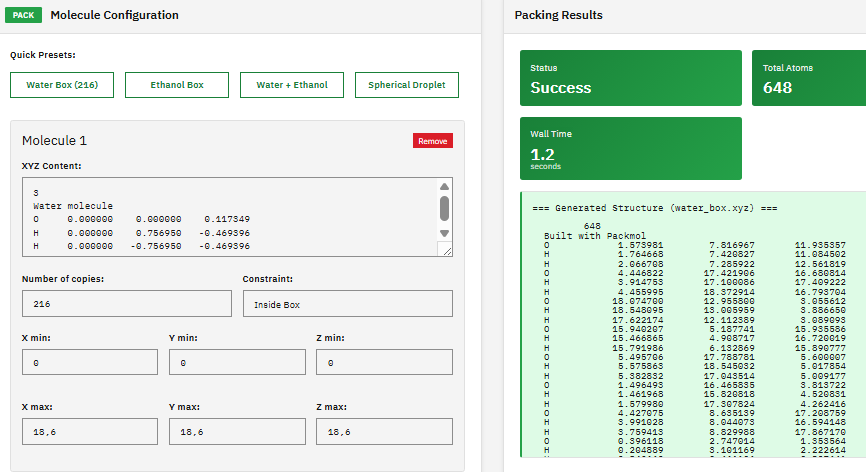
Packmol
MIT (Free)
A molecular packing tool that creates initial configurations for molecular dynamics simulations by positioning molecules into defined geometric regions (boxes, spheres, cylinders) while avoiding atomic overlaps and steric clashes.
Features & Details
Key Features
- Geometry: boxes, spheres, cylinders, planes
- Constraints: distance, inside/outside regions
- Output: PDB, XYZ coordinate files
- Integration: GROMACS, NAMD, AMBER workflows
Martinez, L. et al. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157-2164 (2009). DOI:10.1002/jcc.21224
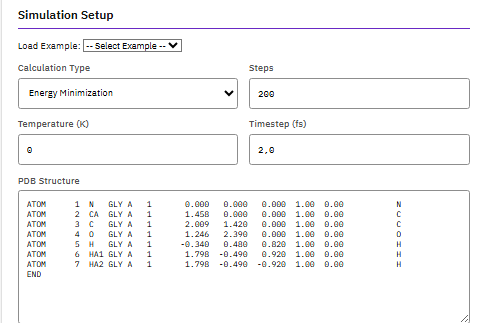
NAMD
Nanoscale Molecular Dynamics
UIUC-NAMD (Partial)
A parallel molecular dynamics engine for simulating large biomolecular systems, supporting CHARMM, AMBER, and OPLS force fields with free energy perturbation, replica exchange, and QM/MM capabilities.
Features & Details
Key Features
- Force fields: CHARMM, AMBER, OPLS
- Methods: FEP, replica exchange, QM/MM
- Scalability: MPI, GPU acceleration
- Integration: VMD visualization
Phillips, J.C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153, 044130 (2020). DOI:10.1063/5.0014475
Free for academic use. Commercial license required from UIUC.
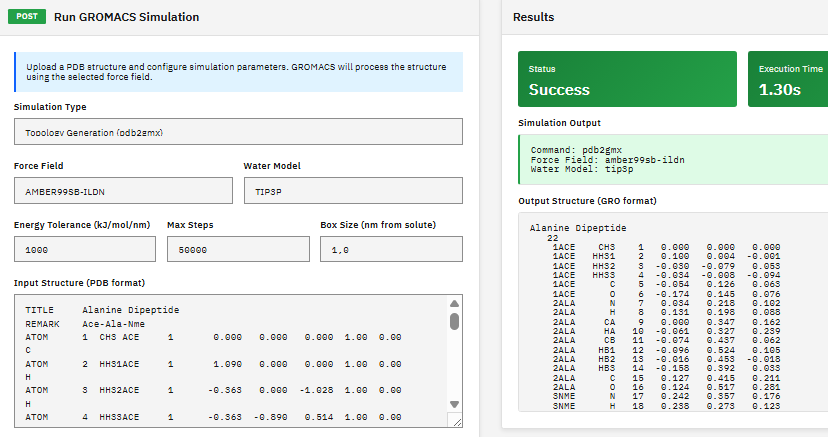
GROMACS
LGPL-2.1 (Free)
A molecular dynamics package for simulating proteins, lipids, and nucleic acids, with multi-level parallelism supporting CUDA GPU acceleration, MPI distributed computing, and OpenMP threading.
Features & Details
Key Features
- Force fields: AMBER, CHARMM, OPLS, GROMOS
- GPU: CUDA, SYCL acceleration
- Analysis: g_rms, g_energy, g_rdf tools
- Free energy: TI, FEP, BAR methods
Abraham, M.J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19-25 (2015). DOI:10.1016/j.softx.2015.06.001
GAMESS
General Atomic and Molecular Electronic Structure System
Proprietary Academic (Partial)
An ab initio quantum chemistry package supporting Hartree-Fock, DFT, MP2, coupled cluster, MCSCF, and CI methods for electronic structure calculations, geometry optimizations, and property predictions.
Features & Details
Key Features
- Methods: HF, DFT, MP2, CCSD, MCSCF, CI
- Basis sets: 6-31G, cc-pVXZ, aug-cc-pVXZ
- Properties: NMR, IR, Raman, UV-Vis
- Solvation: PCM, EFP models
Schmidt, M.W. et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 14, 1347-1363 (1993). DOI:10.1002/jcc.540141112
Academic license registration required. Currently on hold.
NWChem
ECL-2.0 (Free)
A computational chemistry platform providing Hartree-Fock, DFT, MP2, and coupled cluster methods with MPI parallelization for electronic structure simulations on computing clusters.
Features & Details
Key Features
- Methods: HF, DFT, MP2, CCSD, CCSD(T)
- Basis sets: Gaussian, plane-wave
- Parallelization: MPI, Global Arrays
- Properties: NMR, gradients, Hessians
Valiev, M. et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 181, 1477-1489 (2010). DOI:10.1016/j.cpc.2010.04.018
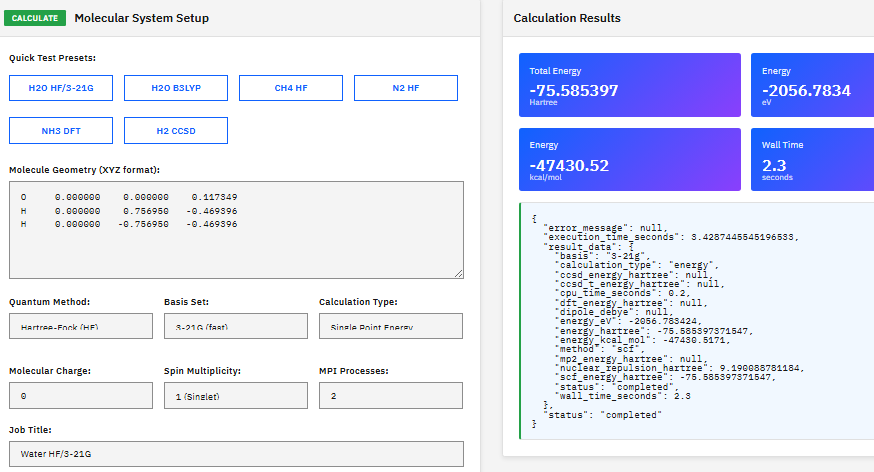
AmberTools
GPL-2.0 (Free)
A force field parameterization and molecular dynamics preparation suite providing antechamber for GAFF/GAFF2 atom typing, tleap for building simulation systems, cpptraj for trajectory analysis, and reduce for hydrogen optimization.
Features & Details
Key Features
- Parameterization: antechamber, parmchk2
- System building: tleap, xleap
- Analysis: cpptraj, pytraj
- Force fields: GAFF, GAFF2, ff19SB
Case, D.A. et al. AmberTools. J. Chem. Inf. Model. 63, 6183-6191 (2023). DOI:10.1021/acs.jcim.3c01153
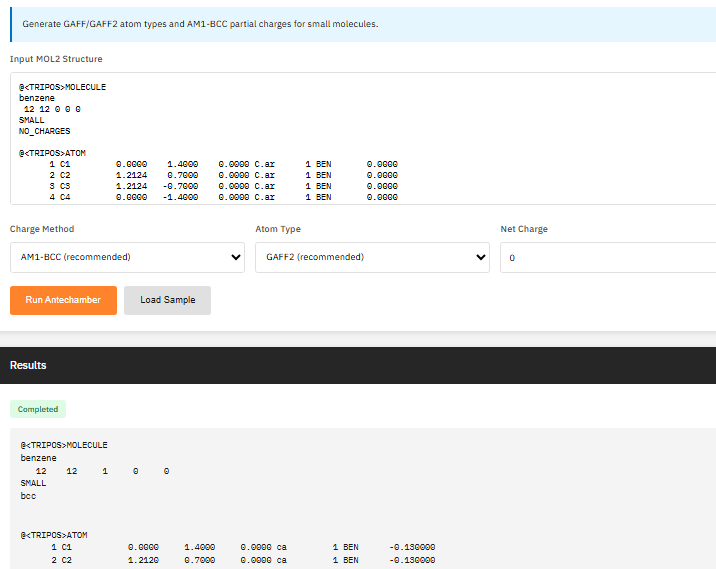
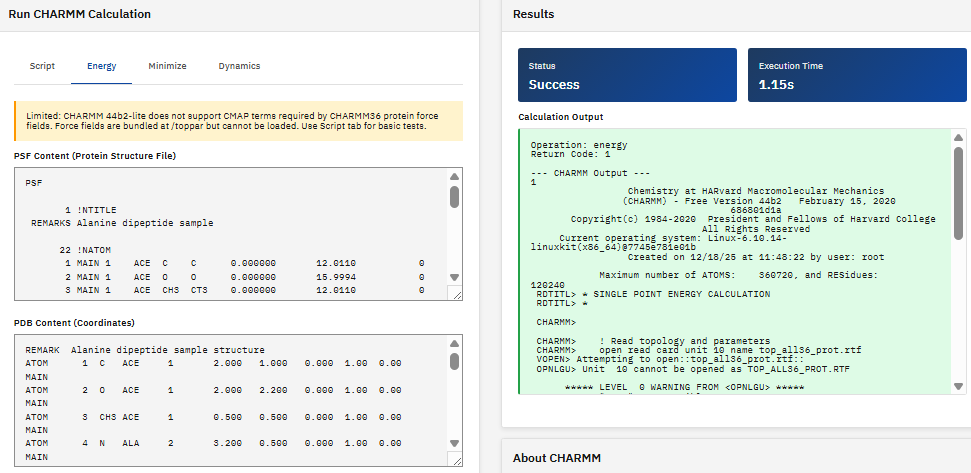
CHARMM
Chemistry at HARvard Macromolecular Mechanics
Academic/Non-profit (Partial)
A molecular simulation program for modeling macromolecular systems, providing energy calculations, minimization, dynamics simulations, and sampling methods for proteins, nucleic acids, lipids, and small molecules.
Features & Details
Key Features
- Force fields: CHARMM36, CGenFF
- Sampling: replica exchange, metadynamics
- QM/MM: interface with ORCA, Gaussian
- Analysis: MMTSB, CHARMM-GUI
Brooks, B.R. et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 30, 1545-1614 (2009). DOI:10.1002/jcc.21287
CHARMm is the commercial version available through Biovia.
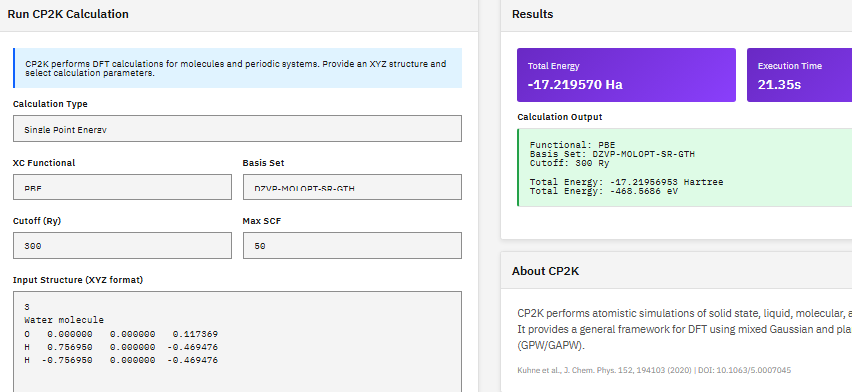
CP2K
GPL-2.0 (Free)
A quantum chemistry and solid-state physics package combining DFT with Gaussian and plane-wave (GPW/GAPW) methods, enabling geometry optimization, cell optimization, and ab initio molecular dynamics for both periodic crystals and isolated molecular systems.
Features & Details
Key Features
- Methods: DFT, HF, MP2, RPA
- Basis: GPW, GAPW mixed Gaussian/plane-wave
- MD: Born-Oppenheimer, Car-Parrinello
- Parallelization: MPI, OpenMP, GPU
Kuhne, T.D. et al. CP2K: An electronic structure and molecular dynamics software package. J. Chem. Phys. 152, 194103 (2020). DOI:10.1063/5.0007045
ORCA
Academic free / commercial license (Partial)
A versatile quantum chemistry program supporting DFT, ab initio, and semi-empirical methods. ORCA enables accurate calculations of molecular structures, spectra, and reaction mechanisms and serves as a backend for AI-assisted computational chemistry workflows.
Features & Details
Key Features
- Methods: DFT, HF, MP2, CCSD(T), CASSCF
- Spectroscopy: NMR, EPR, UV-Vis, IR, Raman
- Solvation: CPCM, SMD models
- Parallelization: MPI, OpenMP
Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 12, e1606 (2022). DOI:10.1002/wcms.1606
Free for academic use. Commercial license via FACCTs GmbH.
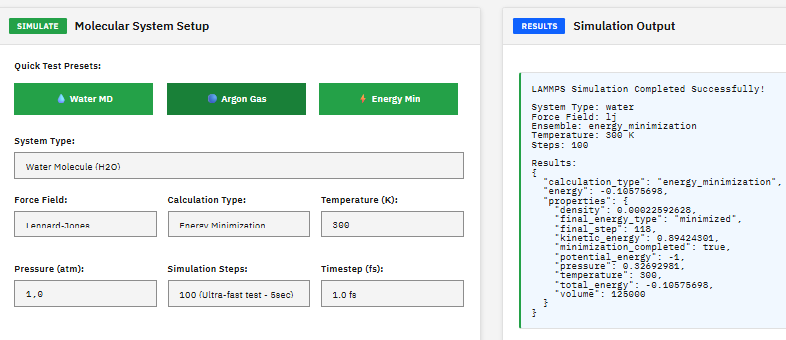
LAMMPS
Large-scale Atomic/Molecular Massively Parallel Simulator
Academic / non-commercial (Partial)
A molecular dynamics engine for scalable simulations of materials and polymers. Supports a wide variety of force fields, potentials, and simulation methods.
Features & Details
Key Features
- Potentials: LJ, EAM, Tersoff, ReaxFF, AIREBO
- Ensembles: NVE, NVT, NPT, NPH
- GPU: CUDA, OpenCL, Kokkos
- Parallelization: MPI, OpenMP
Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 117, 1-19 (1995). DOI:10.1006/jcph.1995.1039
Academic/non-commercial license applies.
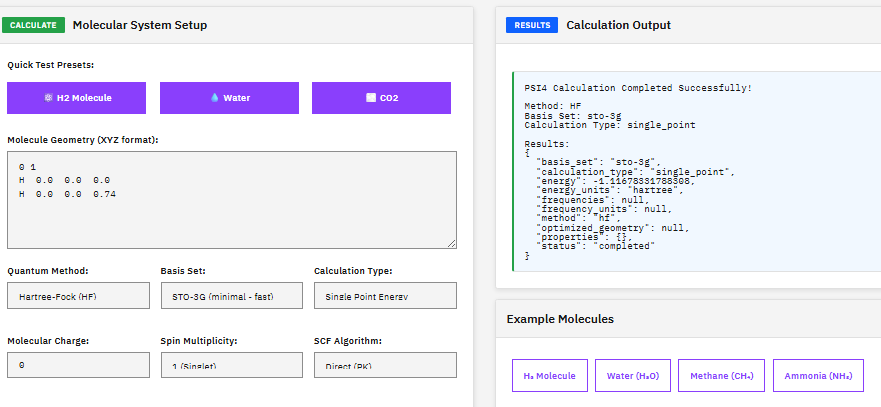
PSI4
LGPL-3.0 (Free)
A quantum chemistry suite providing Hartree-Fock, DFT, MP2, coupled cluster, and symmetry-adapted perturbation theory (SAPT) methods for high-throughput electronic structure calculations.
Features & Details
Key Features
- Methods: HF, DFT, MP2, CCSD(T), SAPT
- Basis: cc-pVXZ, aug-cc-pVXZ, def2
- Properties: gradients, Hessians, NMR
- Integration: Python API, NumPy arrays
Smith, D.G.A. et al. Psi4 1.4: Open-Source Software for High-Throughput Quantum Chemistry. J. Chem. Phys. 152, 184108 (2020). DOI:10.1063/5.0006002
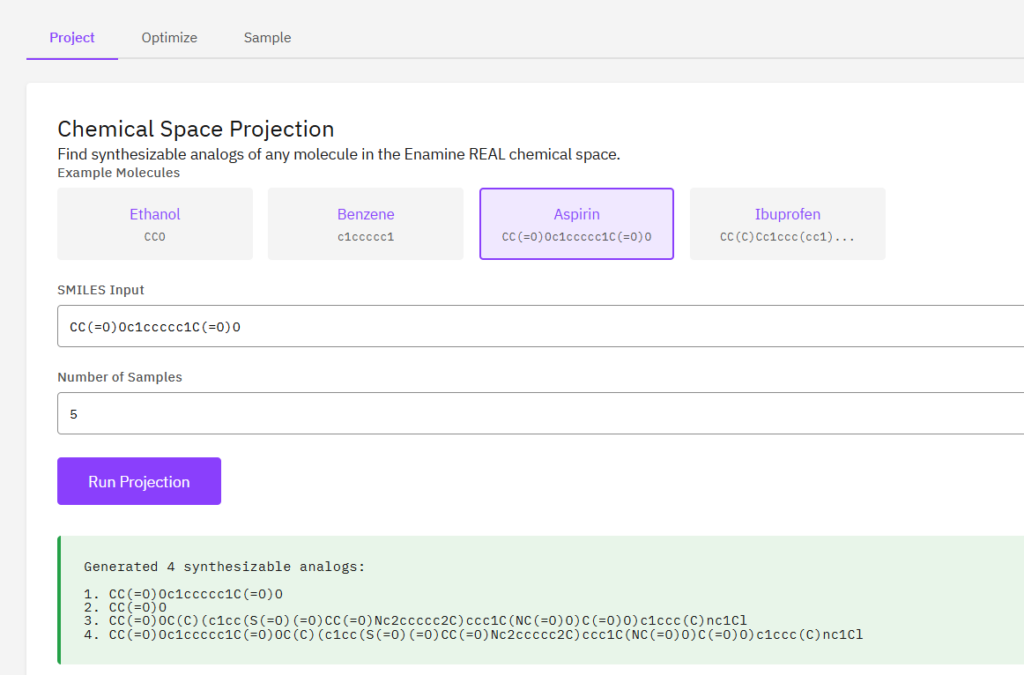
PrexSyn
MIT Academic (Partial)
A synthesizability-constrained generative molecular design framework using a GPT-style decoder-only transformer. Enables projection of any molecule into the Enamine REAL chemical space to find synthesizable analogs from a space of 40B+ molecules.
Features & Details
Luo, S. & Coley, C.W. Synthesizability-Constrained Generative Molecular Design. arXiv:2512.00384 (2024). DOI:10.48550/arXiv.2512.00384
Academic license from MIT. Commercial use requires separate agreement.
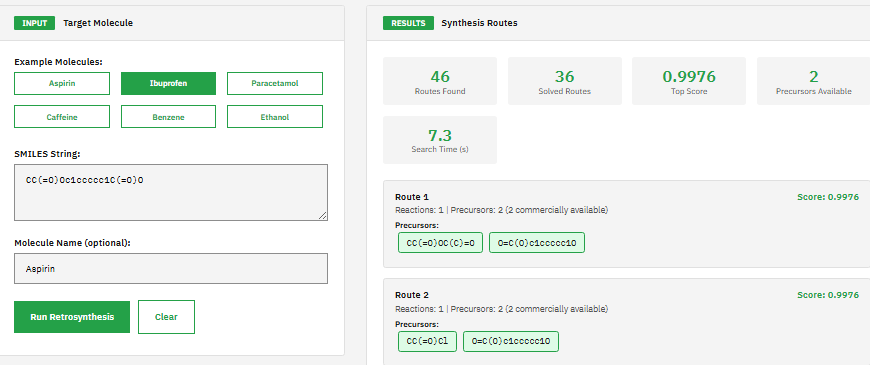
AiZynthFinder
MIT (Free)
AI-driven retrosynthetic route planning tool for computer-aided synthesis using Monte Carlo tree search and deep neural networks to predict multi-step synthesis routes for organic molecules with route scoring and ranking capabilities.
Features & Details
Key Features
- Algorithm: MCTS, neural network policies
- Routes: multi-step retrosynthesis
- Scoring: route feasibility ranking
- Integration: RDKit, PyTorch
Genheden, S. et al. AiZynthFinder: a fast, robust and flexible open-source software for retrosynthetic planning. J. Cheminform. 12, 70 (2020). DOI:10.1186/s13321-020-00472-1
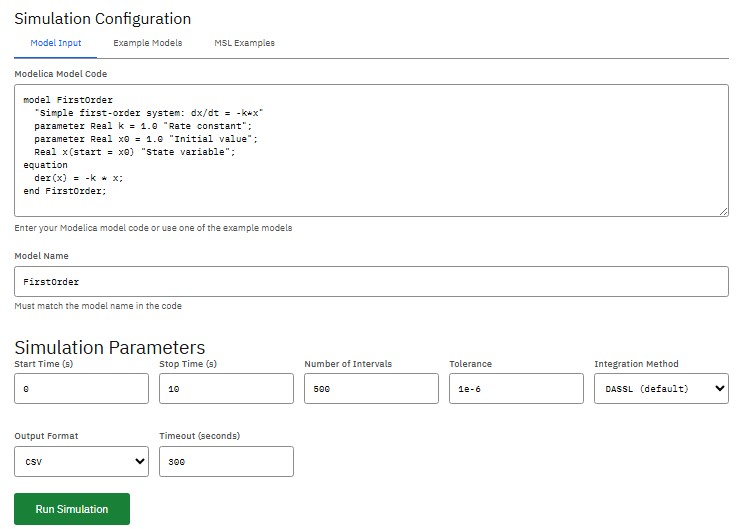
OpenModelica
OSMC Public License (Free)
Simulation environment based on the Modelica language, for industrial and academic use in developing complex cyber-physical systems and analyzing multi-domain systems governed by differential, algebraic, and discrete equations.
Features & Details
Key Features
- Language: Modelica 3.5+ standard
- Domains: mechanical, electrical, thermal, fluid
- Tools: OMEdit GUI, OMShell, OMNotebook
- Export: FMI 2.0 co-simulation
Fritzson, P. et al. The OpenModelica Integrated Environment for Modeling, Simulation, and Model-Based Development. Modeling, Identification and Control. 41(4), 241-295 (2020). DOI:10.4173/mic.2020.4.1
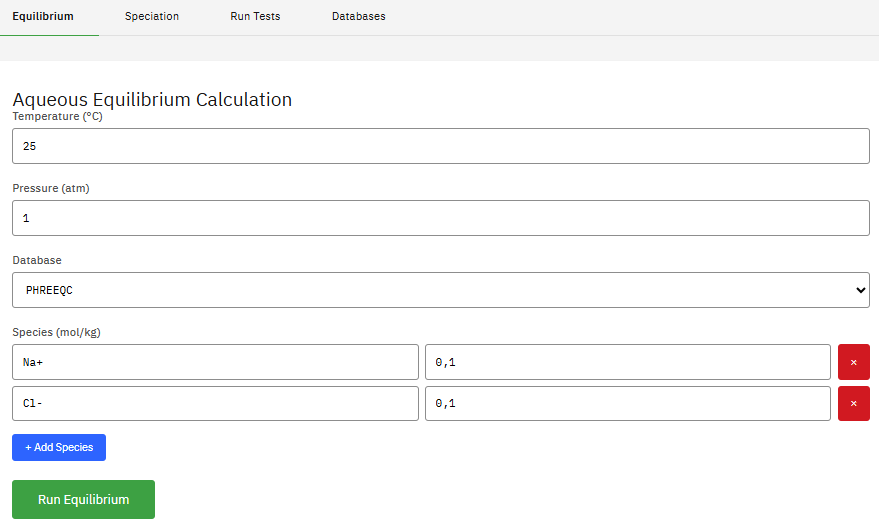
Reaktoro
LGPL-2.1 (Free)
Modeling complex chemically reactive systems governed by chemical equilibrium, kinetics, or a combination. Efficient numerical algorithms like Gibbs energy minimization and on-demand machine learning for simulations.
Features & Details
Key Features
- Equilibrium: Gibbs energy minimization
- Kinetics: reaction rate equations
- Databases: SUPCRT, PHREEQC, ThermoFun
- Applications: geochemistry, CO2 storage
Leal, A.M.M. Reaktoro: An open-source unified framework for modeling chemically reactive systems (2015). https://reaktoro.org
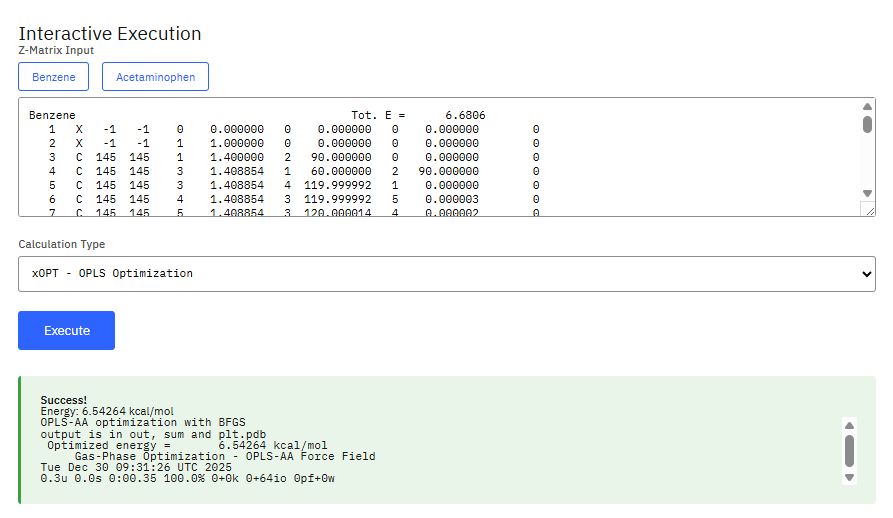
BOSS
Biochemical and Organic Simulation System
Academic free / commercial required (Partial)
Molecular modeling for MM, Metropolis Monte Carlo simulations, and semiempirical quantum mechanics to study organic and biomolecular systems. Used for energy minimizations, conformational searching, and calculating free energies of solvation and binding using the OPLS force fields.
Features & Details
Key Features
- Force fields: OPLS-AA, OPLS/CM1A
- Methods: Monte Carlo, minimization
- Free energy: FEP, TI calculations
- Solvation: explicit, GB/SA continuum
Jorgensen, W.L.; Tirado-Rives, J. Molecular modeling of organic and biomolecular systems using BOSS and MCPRO. J. Comput. Chem. 26, 1689-1700 (2005). DOI:10.1002/jcc.20297
Free for academic use. Commercial license required for for-profit use.
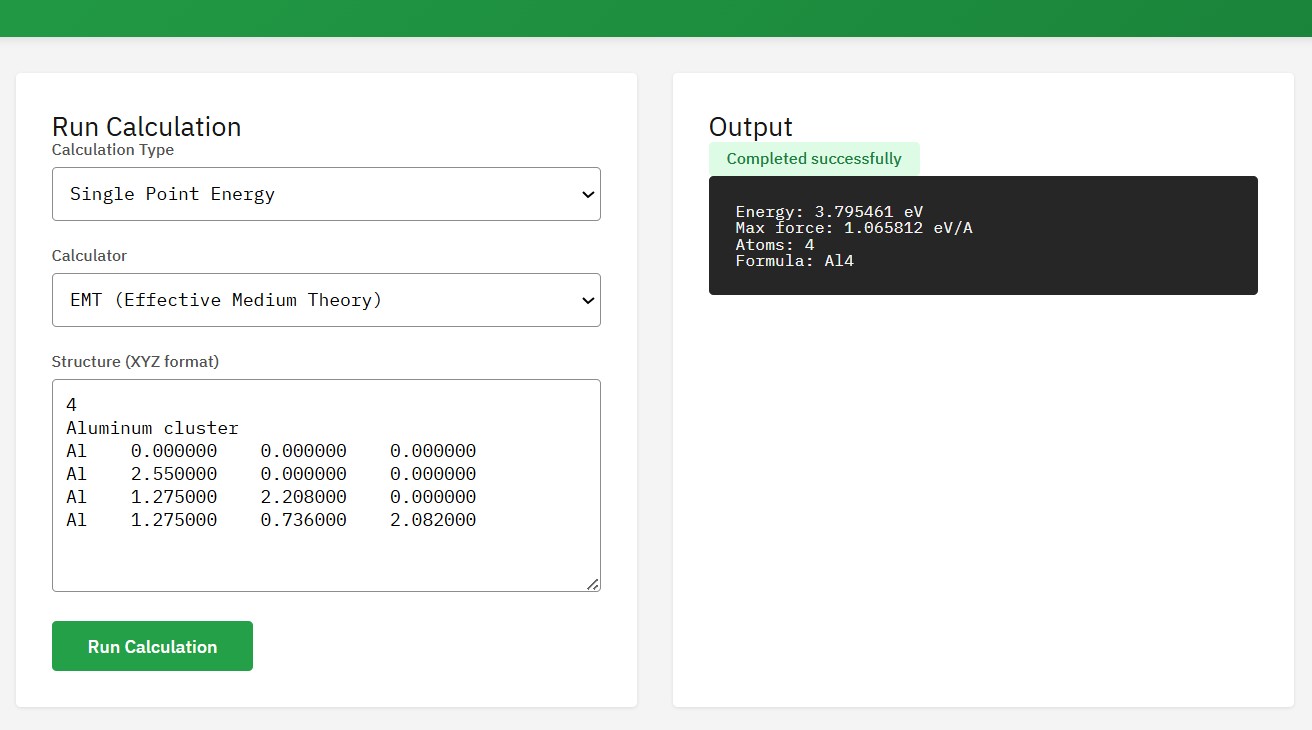
ASE
Atomic Simulation Environment
LGPL-2.1 (Free)
A set of tools for setting up, manipulating, running, visualizing, and analyzing atomistic simulations. It interfaces with many external electronic structure codes (calculators) like VASP, GPAW, Quantum ESPRESSO, and neural network potentials.
Features & Details
Key Features
- Calculators: VASP, GPAW, QE, ORCA, xTB
- Structure: Atoms object, ase.io readers
- Optimization: BFGS, FIRE, LBFGS
- Visualization: ase.visualize, GUI
Larsen, A.H. et al. The Atomic Simulation Environment – A Python library for working with atoms. J. Phys.: Condens. Matter 29, 273002 (2017). DOI:10.1088/1361-648X/aa680e
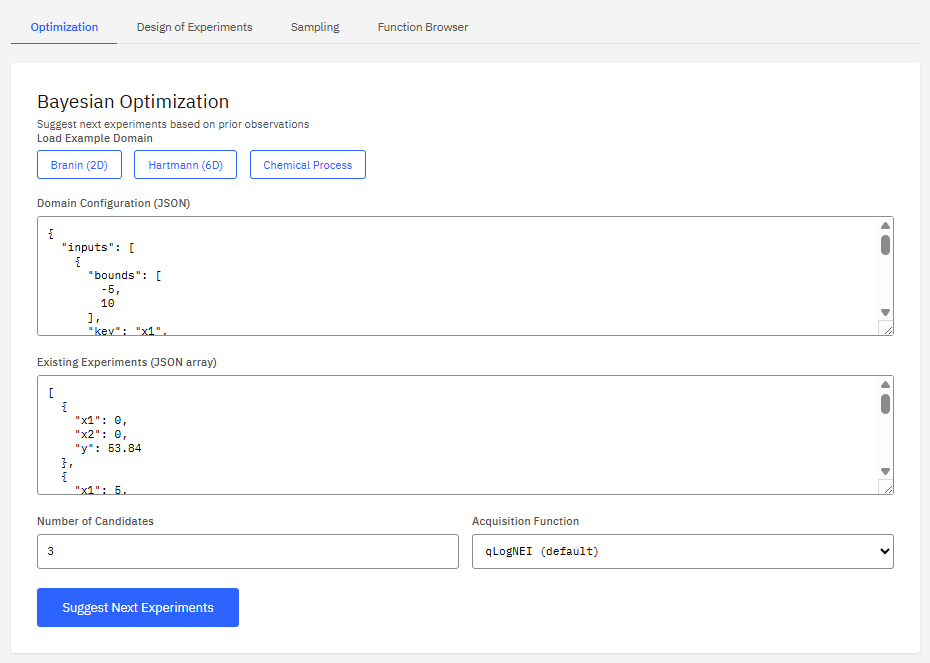
BoFire
Apache-2.0 (Free)
A Bayesian optimization framework designed for experimental design in chemistry and materials science. It optimizes black-box functions with mixed features, constraints, and multi-objective targets, supporting design of experiments (DoE) workflows.
Features & Details
Key Features
- Optimization: Bayesian, multi-objective
- Constraints: linear, nonlinear, discrete
- DoE: Latin hypercube, Sobol sequences
- Integration: BoTorch, GPyTorch backend
BASF Digital Solutions GmbH. BoFire: Bayesian Optimization Framework for Industrial Research and Engineering. https://github.com/experimental-design/bofire
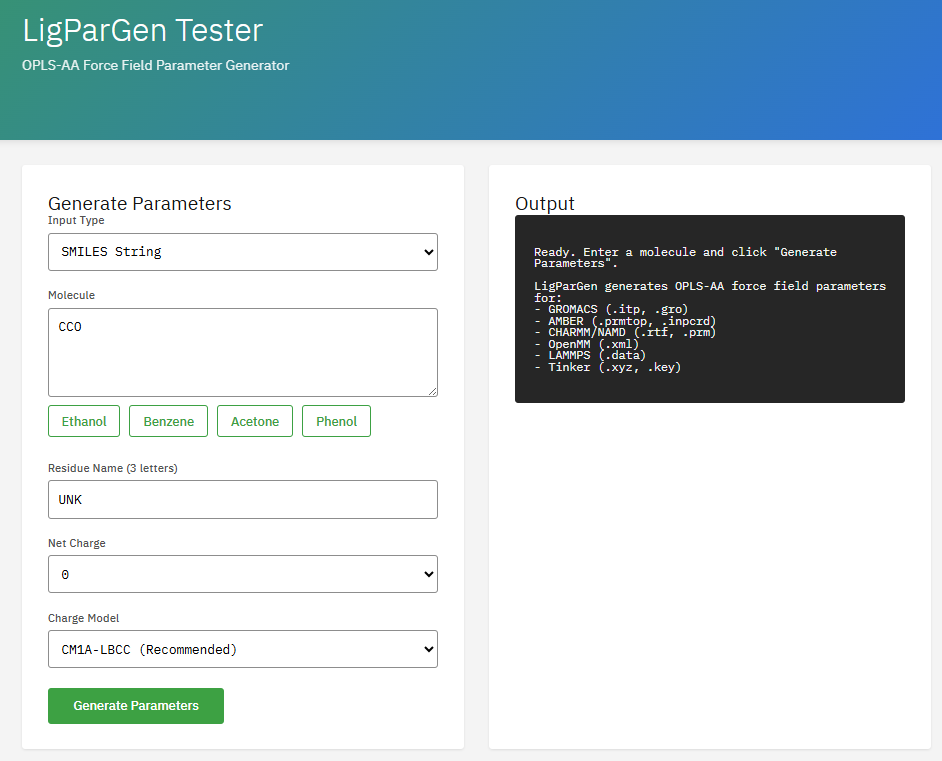
LigParGen
Apache-2.0 (Free)
Program for generating OPLS-AA/1.14*CM1A force field parameters for organic ligands. It creates topology files compatible with major MD engines like GROMACS, CHARMM, OpenMM, and NAMD.
Features & Details
Key Features
- Force field: OPLS-AA, 1.14*CM1A charges
- Output: GROMACS, CHARMM, OpenMM, NAMD
- Input: SMILES, PDB, MOL2
- Charges: CM1A-LBCC, 1.14*CM1A
Dodda, L.S. et al. LigParGen web server: an automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 45, W331-W336 (2017). DOI:10.1093/nar/gkx312
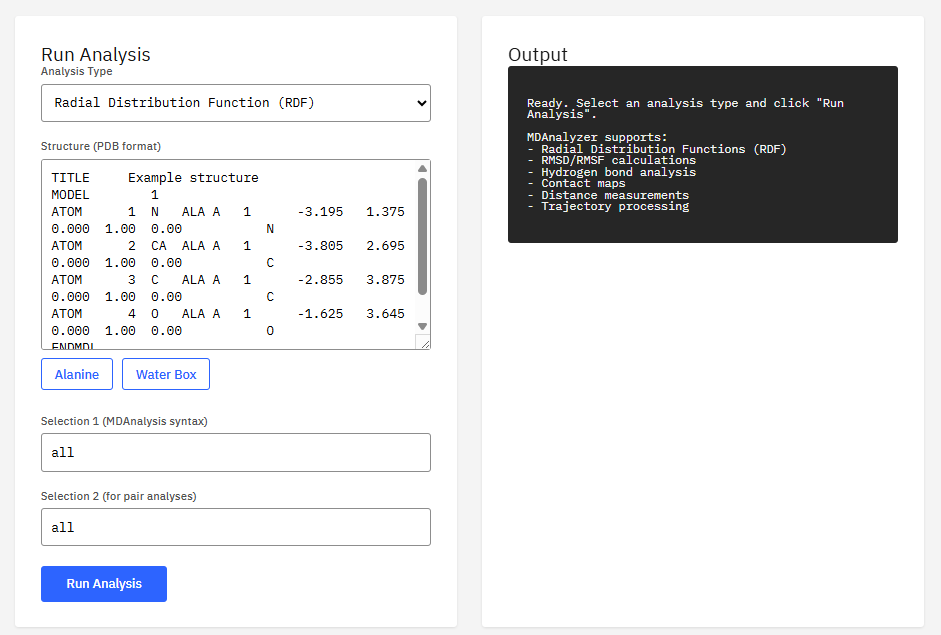
MDAnalysis
GPL-2.0 (Free)
Program to analyze trajectories from molecular dynamics simulations. It supports many file formats (CHARMM, DL_POLY, GROMACS, LAMMPS, NAMD, Amber) and allows for distance calculations, RMSD, and complex analysis.
Features & Details
Key Features
- Formats: DCD, XTC, TRR, NetCDF, PDB
- Analysis: RMSD, RMSF, RDF, contacts
- Selection: MDAnalysis atom selection language
- Integration: NumPy, SciPy, parallel
Michaud-Agrawal, N. et al. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 32, 2319-2327 (2011). DOI:10.1002/jcc.21787
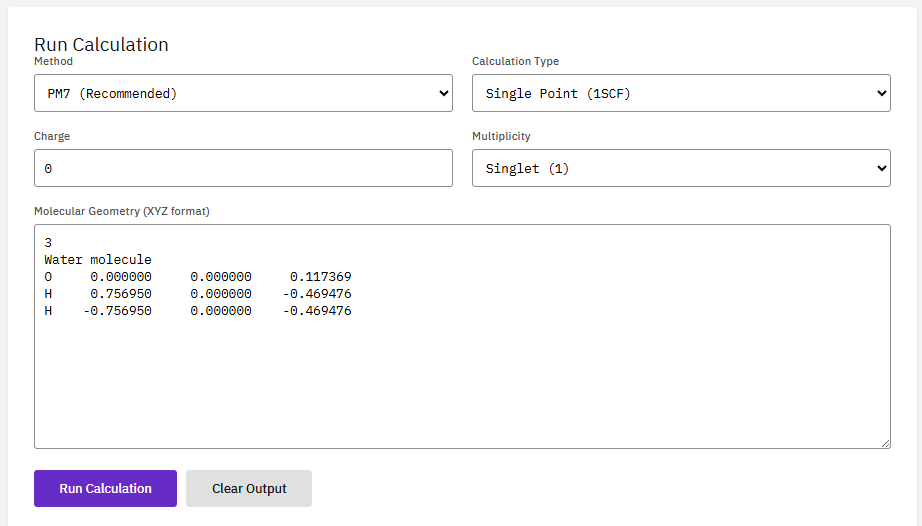
MOPAC
Apache-2.0 (Free)
A semi-empirical quantum chemistry program based on the NDDO approximation. It performs rapid electronic structure calculations, geometry optimizations, and reaction path analysis for much larger systems than ab initio methods can handle.
Features & Details
Key Features
- Methods: PM6, PM7, RM1, AM1, MNDO
- Calculations: geometry, TS, IRC, thermo
- Solvation: COSMO continuum model
- Speed: 1000x faster than DFT
Stewart, J.J.P. Optimization of parameters for semiempirical methods VI. J. Mol. Model. 19, 1-32 (2013). DOI:10.1007/s00894-012-1667-x
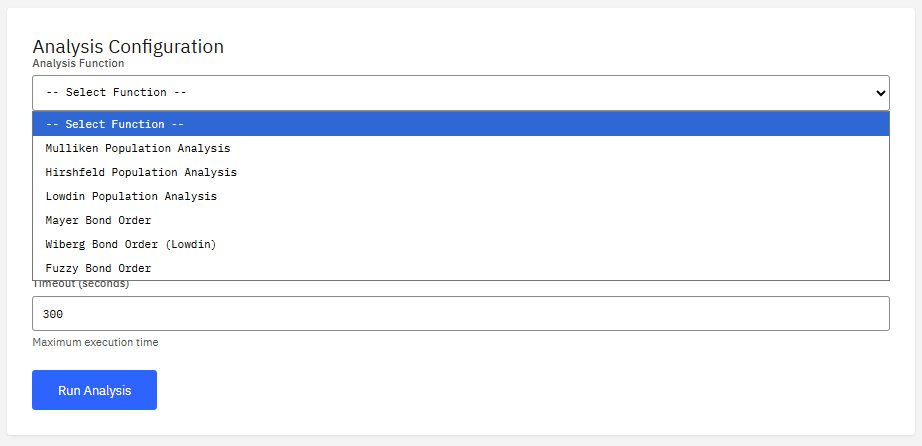
Multiwfn
Freeware (Free)
A wavefunction analysis program. It calculates real space functions, performing population analysis, orbital composition analysis, and plotting various spectra (IR, Raman, UV-Vis) to visualize electronic structure properties.
Features & Details
Key Features
- Analysis: Mulliken, NBO, AIM, ELF, LOL
- Spectra: IR, Raman, UV-Vis, NMR
- Visualization: orbitals, density, ESP
- Input: Gaussian, ORCA, Molden, fchk
Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580-592 (2012). DOI:10.1002/jcc.22885
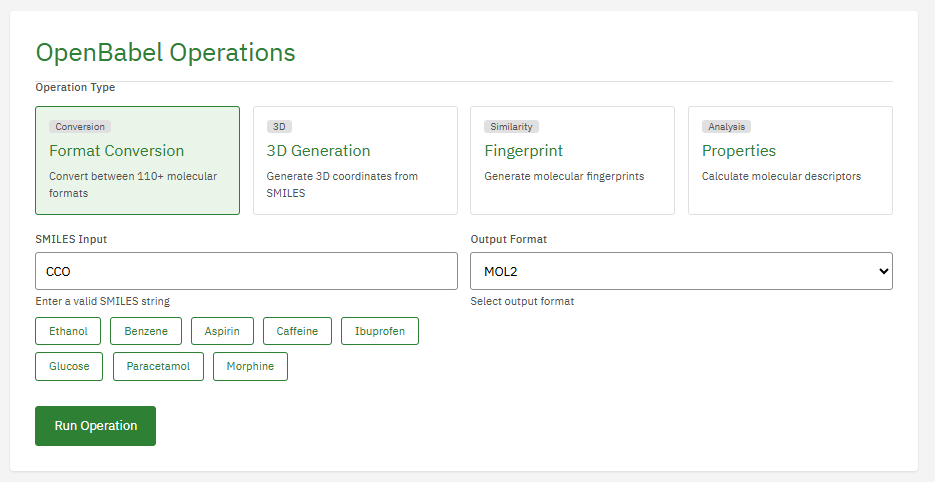
Open Babel
GPL-2.0 (Free)
A chemical toolbox designed to speak the many languages of chemical data. It converts between file formats (SMILES, PDB, Mol2, CML) and provides a library for cheminformatics, substructure searching, and chemical property calculation.
Features & Details
Key Features
- Formats: 110+ chemical file formats
- Conversion: SMILES, InChI, PDB, SDF, MOL2
- Fingerprints: FP2, FP3, FP4, MACCS
- Properties: MW, logP, TPSA, rotatable bonds
O’Boyle, N.M. et al. Open Babel: An open chemical toolbox. J. Cheminform. 3, 33 (2011). DOI:10.1186/1758-2946-3-33
PySCF
Apache-2.0 (Free)
A simulation framework for ab initio electronic structure theory. It supports Hartree-Fock, DFT, Coupled Cluster (CCSD), and CI methods, emphasizing simplicity, extensibility, and efficient handling of large systems.
Features & Details
Key Features
- Methods: HF, DFT, MP2, CCSD, CASSCF, FCI
- Basis: Gaussian basis sets, ECPs
- Periodic: PBC, k-point sampling
- Integration: NumPy, GPU via CuPy
Sun, Q. et al. PySCF: the Python-based simulations of chemistry framework. WIREs Comput. Mol. Sci. 8, e1340 (2018). DOI:10.1002/wcms.1340
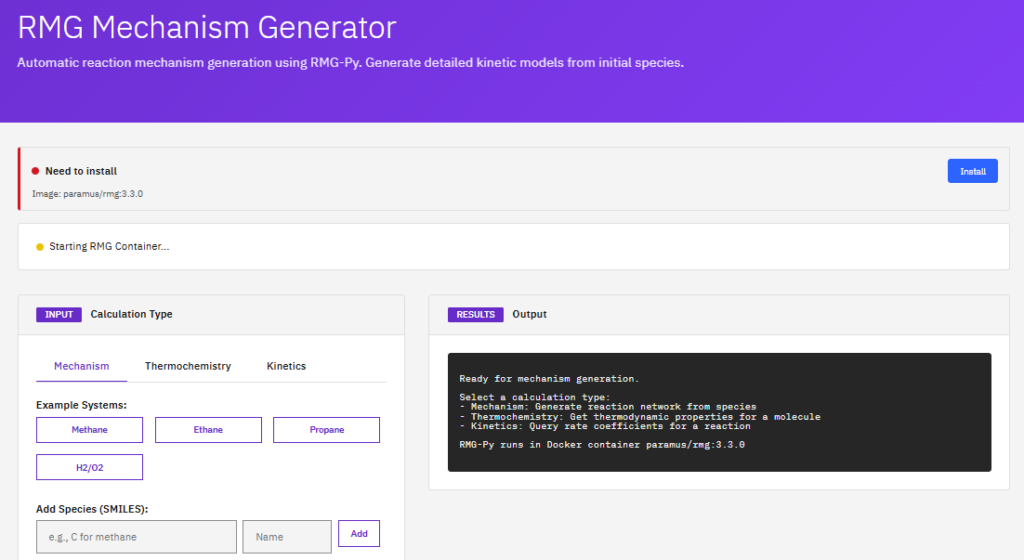
RMG
Reaction Mechanism Generator
MIT (Free)
An automatic chemical reaction mechanism generator that constructs kinetic models composed of elementary chemical reaction steps. It uses thermodynamic estimation and rate rules to predict mechanisms for combustion and atmospheric chemistry.
Features & Details
Key Features
- Mechanisms: automatic generation
- Thermodynamics: group additivity, NASA polynomials
- Kinetics: rate rules, Arrhenius parameters
- Applications: combustion, pyrolysis, atmospheric
Gao, C.W. et al. Reaction Mechanism Generator: Automatic construction of chemical kinetic mechanisms. Comput. Phys. Commun. 203, 212-225 (2016). DOI:10.1016/j.cpc.2016.02.013
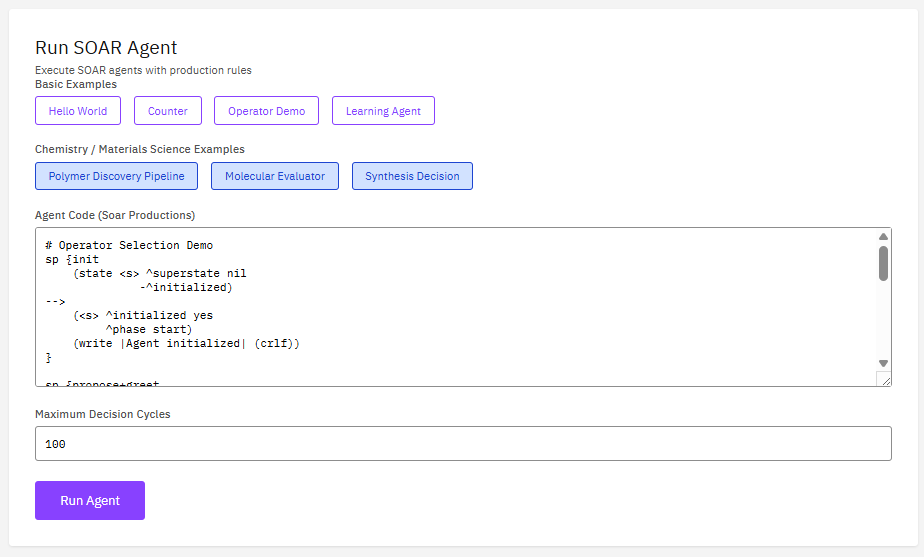
Soar
BSD-3-Clause (Free)
A cognitive architecture for developing systems that exhibit general intelligent behavior. Soar integrates knowledge, planning, learning, and decision-making for agent-based AI modeling and autonomous system development.
Features & Details
Key Features
- Architecture: production rules, working memory
- Learning: chunking, reinforcement learning
- Planning: hierarchical task decomposition
- Integration: external interfaces, SML API
Laird, J.E. The Soar Cognitive Architecture. MIT Press (2012). ISBN:978-0262122962
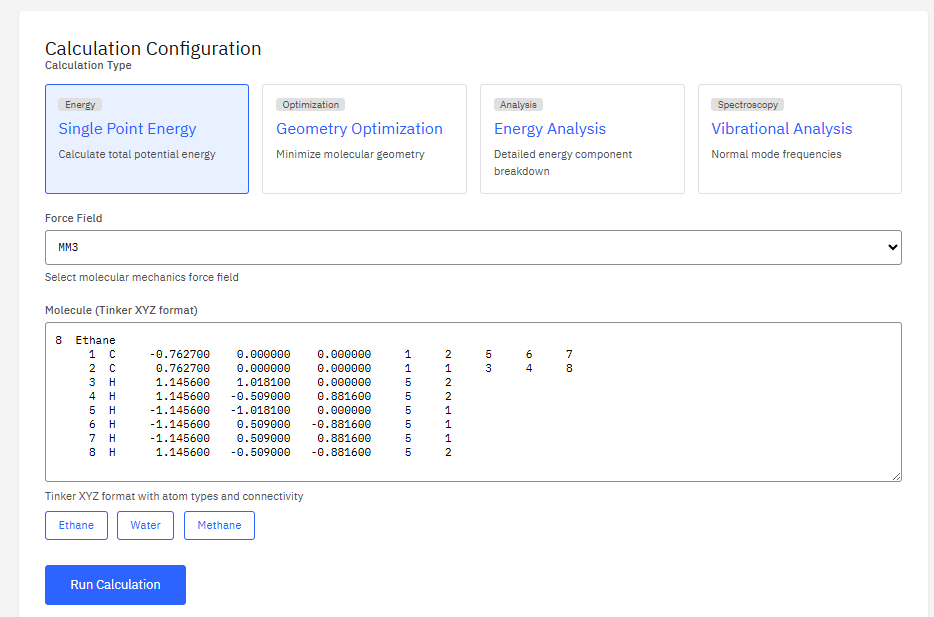
Tinker
Tinker License – academic free (Partial)
A package for molecular mechanics and dynamics, known for its support of advanced polarizable force fields (AMOEBA). It provides tools for geometry optimization, molecular dynamics, and free energy calculations.
Features & Details
Key Features
- Force fields: AMOEBA, OPLS, MM3, CHARMM
- Polarization: induced dipole, Drude
- Methods: minimize, dynamics, analyze
- Free energy: BAR, thermodynamic integration
Rackers, J.A. et al. Tinker 8: Software Tools for Molecular Design. J. Chem. Theory Comput. 14, 5273-5289 (2018). DOI:10.1021/acs.jctc.8b00529
Free for academic/non-profit. Commercial license required.
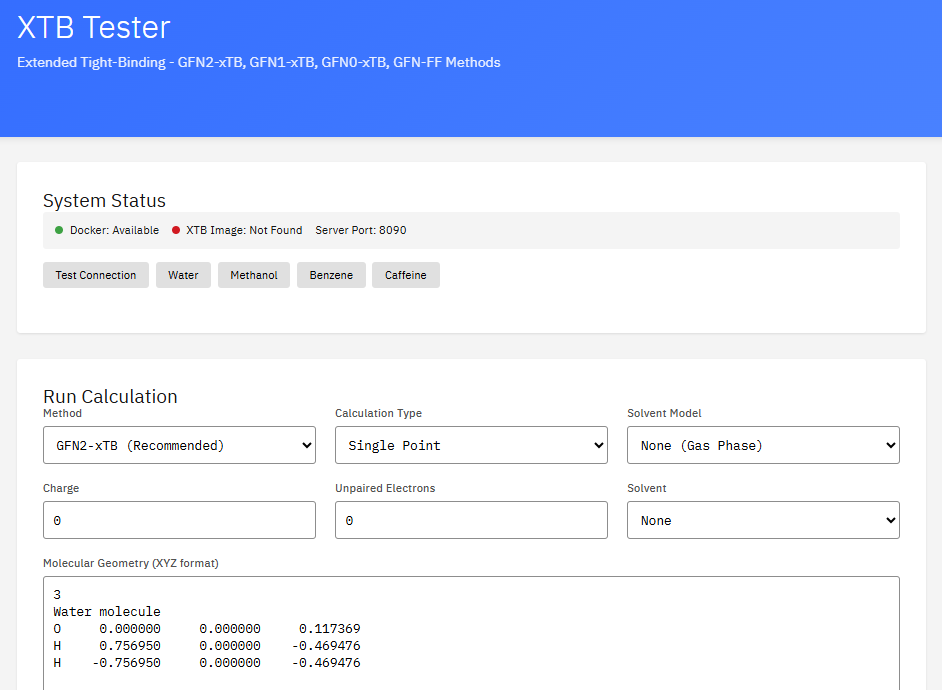
xTB
Extended Tight Binding
LGPL-3.0 (Free)
A semi-empirical quantum chemical program suite developed by the Grimme group. It performs fast and accurate calculations of structures, energies, and properties using the GFN-xTB family of methods for large molecular systems.
Features & Details
Key Features
- Methods: GFN0-xTB, GFN1-xTB, GFN2-xTB
- Speed: 1000x faster than DFT
- Applications: conformers, reactions, MD
- Properties: energies, gradients, Hessians
Bannwarth, C. et al. GFN2-xTB – An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method. J. Chem. Theory Comput. 15, 1652-1671 (2019). DOI:10.1021/acs.jctc.8b01176
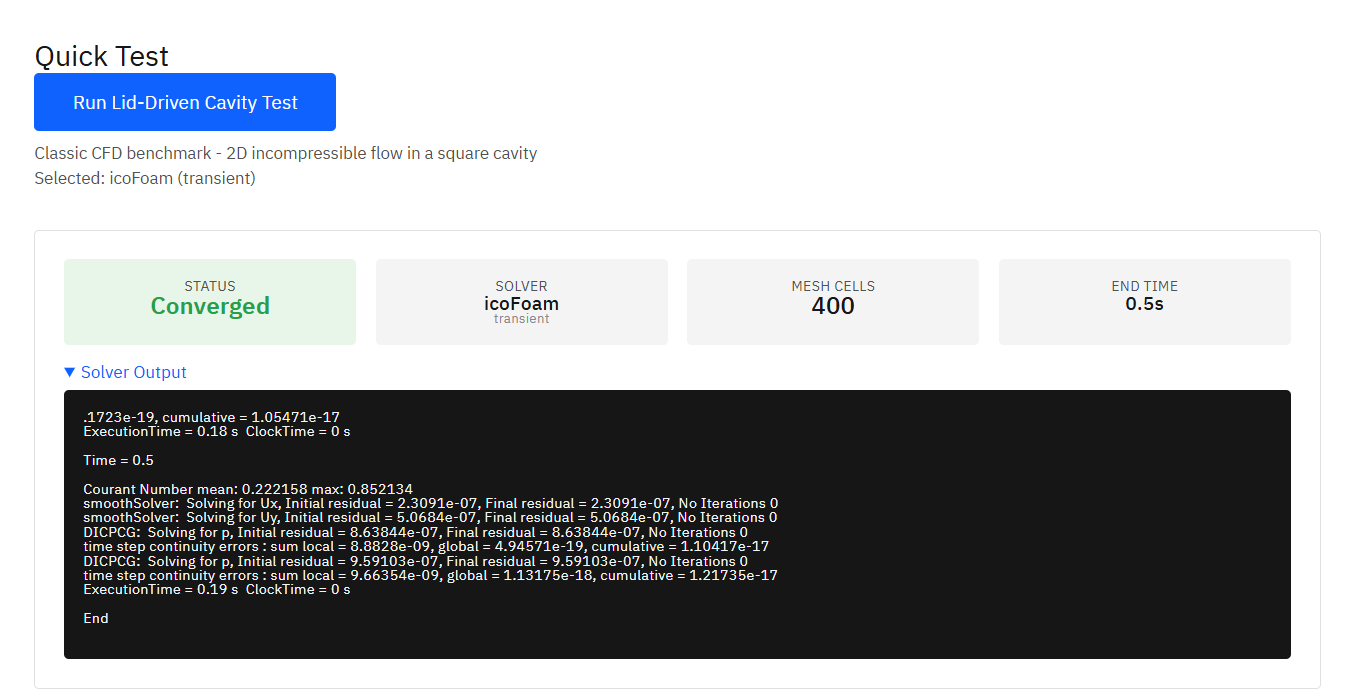
OpenFOAM
GPL-3.0 (Free)
A computational fluid dynamics (CFD) toolbox for complex fluid flows including incompressible and compressible flow, turbulence modeling, multiphase flows, and heat transfer. Supports both transient and steady-state simulations.
Features & Details
Weller, H.G. et al. A tensorial approach to computational continuum mechanics using object-oriented techniques. Computers in Physics. 12, 620-631 (1998). DOI:10.1063/1.168744
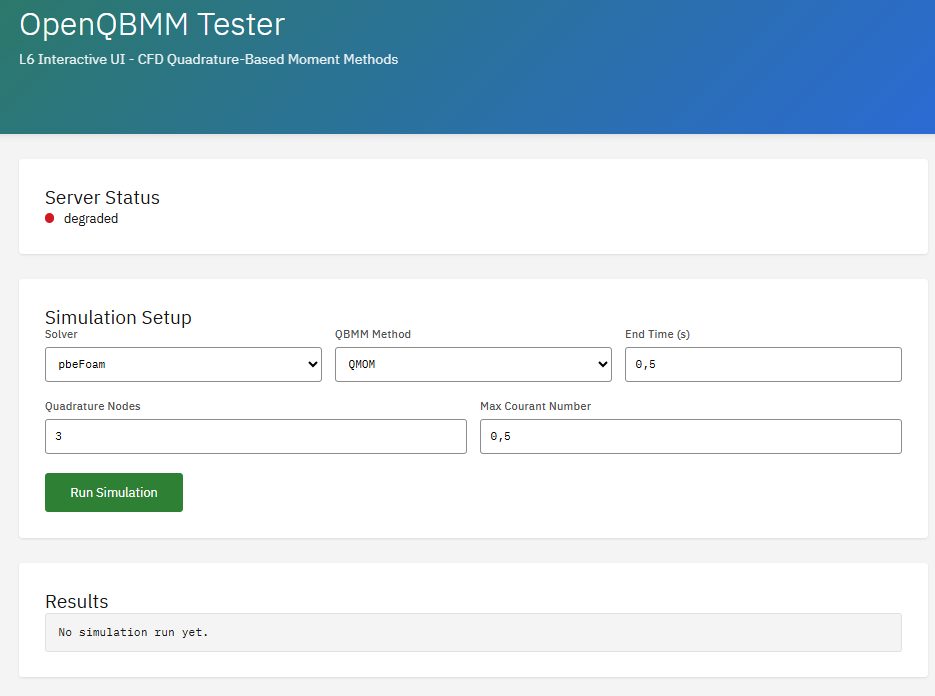
OpenQBMM
GPL-3.0 (Free)
A solver suite for polydisperse multiphase flows using Quadrature-Based Moment Methods (QBMM). Requires OpenFOAM as base platform. Enables simulation of population balance equations, particle size distributions, spray atomization, and aerosol dynamics.
Features & Details
Passalacqua, A. et al. An open-source quadrature-based population balance solver for OpenFOAM. Chemical Engineering Science. 176, 306-318 (2018). DOI:10.1016/j.ces.2017.10.043
DWSIM
GPL-3.0 (Free)
Open-source CAPE-OPEN compliant chemical process simulator for steady-state and dynamic simulations. Supports flash calculations (VLE/LLE/VLLE), thermodynamic property packages (Peng-Robinson, SRK, NRTL, UNIQUAC), and unit operations.
Features & Details
Wagner, D. “DWSIM – An Open-Source Chemical Process Simulator” https://dwsim.org/
Code_Saturne
GPL-2.0 (Free)
Open-source CFD software developed by EDF for solving Navier-Stokes equations in 2D/3D flows. Handles steady/unsteady, laminar/turbulent, incompressible/dilatable flows with advanced turbulence modeling, conjugate heat transfer, and multiphase capabilities.
Features & Details
Key Features
- Turbulence: k-epsilon, k-omega, RSM, LES
- Heat transfer: conduction, convection, radiation
- Multiphase: Euler-Euler, Lagrangian tracking
- Mesh: MED, CGNS, Ensight, I-deas formats
Archambeau, F. et al. Code_Saturne: A Finite Volume Code for the computation of turbulent incompressible flows. International Journal on Finite Volumes. 1(1), 1-62 (2004).
Language Models (LLM) (3)
Language Models (LLM)
Running large language models locally is a statement of independence: data stays private, inference remains under full control, and chemistry does not leave the laboratory.
The price is: speed. Today it is painfully slow. But it works.
Ether0:q2_k
Apache-2.0 (Free)
Scientific research language model fine-tuned for chemistry, biology, and materials science. Based on Qwen2.5-24B with Q2_K quantization optimized for single-CPU execution, enabling research workflows on standard hardware.
Features & Details
Narayanan, S.M. et al. Training a Scientific Reasoning Model for Chemistry. arXiv:2506.17238 (2025). DOI:10.48550/arXiv.2506.17238
Llama 3.2:1b
Llama Community License (Partial)
Provides lightweight natural language understanding and generation suitable for embedded and edge devices. Its compact size enables efficient inference on consumer-grade GPUs and even high-performance CPUs. Supports multilingual text processing, basic reasoning, and customizable fine-tuning.
Features & Details
Meta AI. Llama 3.2 1B (1.23-billion-parameter multilingual language model), released 25 September 2024, https://huggingface.co/meta-llama/Llama-3.2-1B
Commercial use allowed under attribution terms (not purchase).
DeepSeek R1:8b
MIT (Free)
A reasoning-focused large language model for scientific and technical tasks, offering chain-of-thought reasoning and step-by-step problem decomposition for general questions.
Features & Details
DeepSeek-AI, Guo, D., Yang, D., Zhang, H., et al. DeepSeek-R1: Incentivizing Reasoning Capability in LLMs via Reinforcement Learning. arXiv:2501.12948 (2025). DOI:10.48550/arXiv.2501.12948
AI Models (8)
AI Models
These models are one-click installable programs following the Paramus AI Runtime Specification.
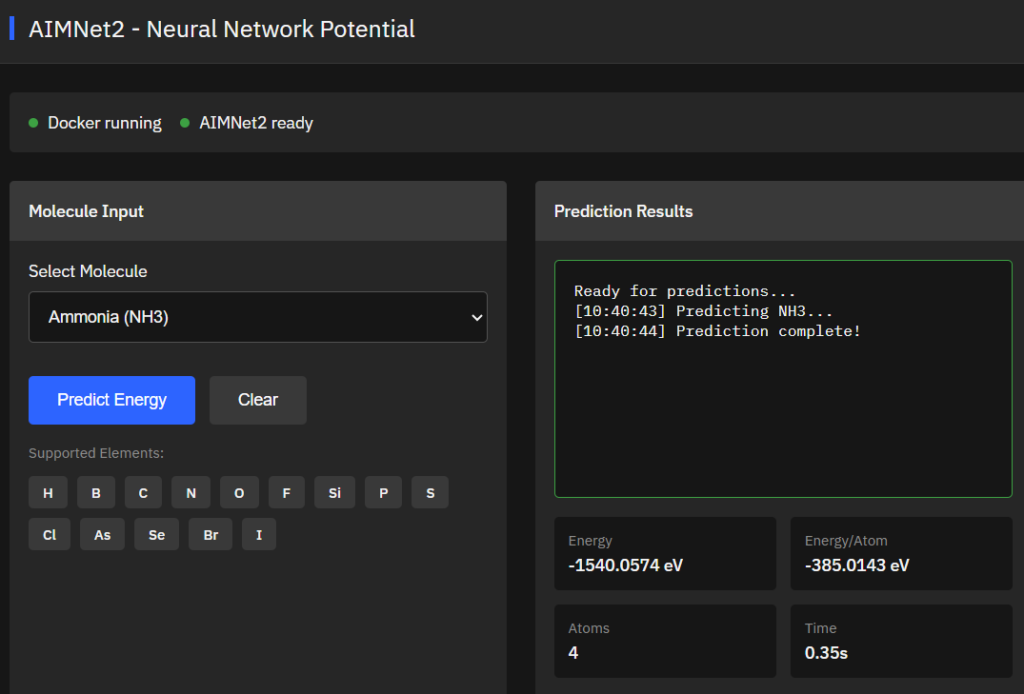
AIMNet2
MIT (Free)
High-accuracy neural network potential for organic and elemental-organic molecules supporting neutral and charged species. Predicts energies, forces, and partial charges at wB97M-D3BJ/def2-TZVPP level for drug discovery and computational chemistry applications.
Features & Details
Anstine, D.M.; Isayev, O. AIMNet2: A Neural Network Potential to Meet your Neutral, Charged, Organic, and Elemental-Organic Needs. J. Phys. Chem. A (2023). DOI:10.1021/acs.jpca.2c06685
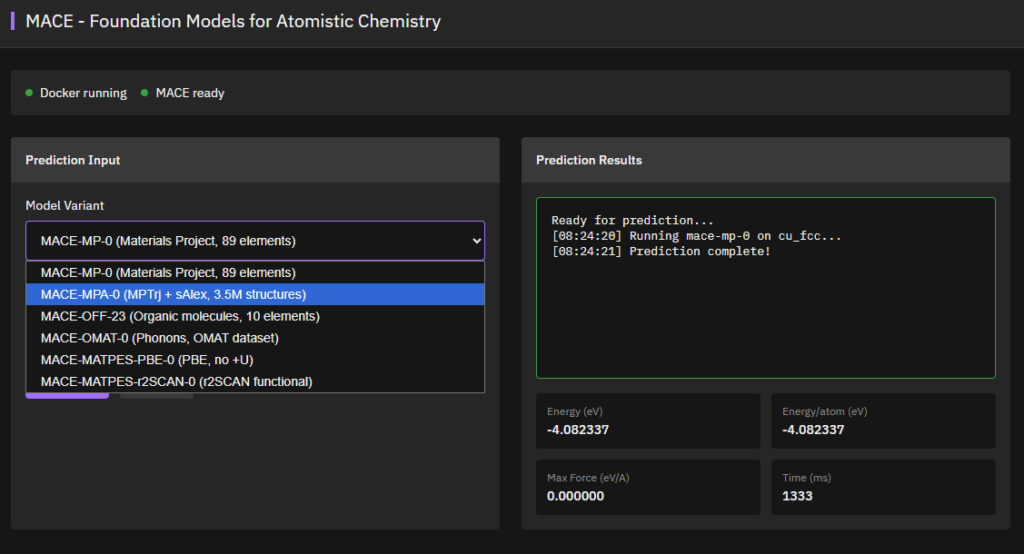
MACE
Apache-2.0 (Free)
Equivariant message passing neural networks achieving state-of-the-art accuracy for atomistic simulations. Universal foundation models for molecular dynamics and materials science with DFT-level accuracy and computational efficiency.
Features & Details
Batatia, I. et al. MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields. NeurIPS (2022). DOI:10.48550/arXiv.2206.07697
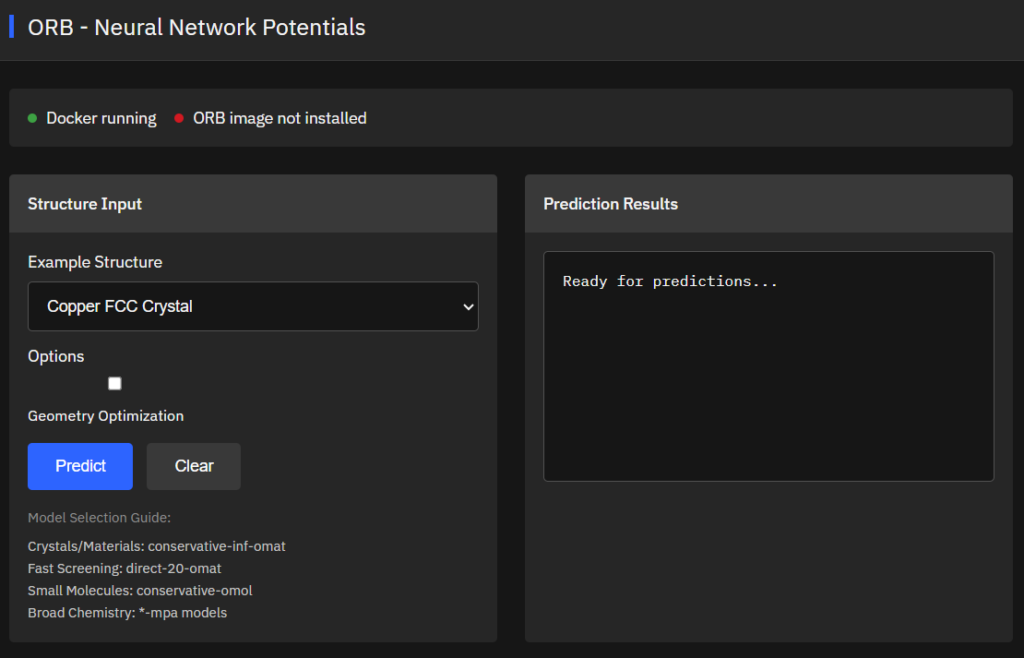
ORB
Apache-2.0 (Free)
Neural network potentials for atomic simulations providing fast and accurate energy/force predictions for materials science, chemistry, and molecular systems. Force fields supporting structure optimization, molecular dynamics, and property calculations.
Features & Details
Rhodes, B. et al. Orb-v3: atomistic simulation at scale. arXiv:2504.06231 (2025).
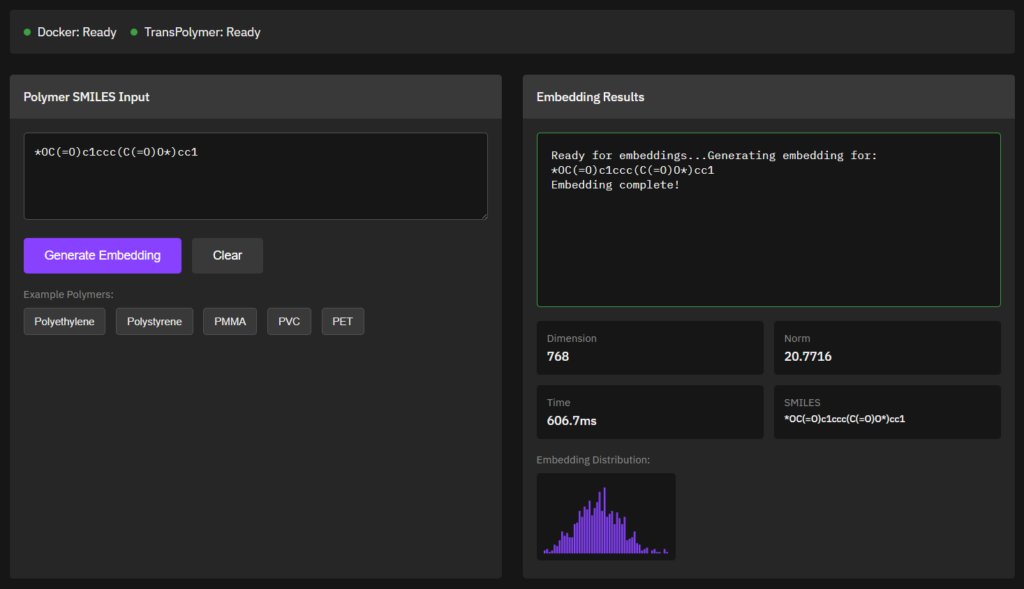
TransPolymer
MIT (Free)
Predicts polymer properties using transformer-based deep learning models trained on polymer structure-property datasets. Designed for inverse design and polymer informatics workflows.
Features & Details
Xu, C.; Wang, Y.; Barati Farimani, A. TransPolymer: a Transformer-based language model for polymer property predictions. npj Computational Materials 9, 64 (2023). DOI:10.1038/s41524-023-01009-9
PolyNC
Apache-2.0 (Free)
A unified natural & chemical language model (text-to-text) for predicting polymer properties (multi-task: regression + classification).
Features & Details
Qiu, H.; Liu, L.; Qiu, X.; Dai, X.; Ji, X.; Sun, Z.-Y. PolyNC: a natural and chemical language model for unified polymer properties prediction. Chemical Science (2024). DOI:10.1039/D3SC05079C
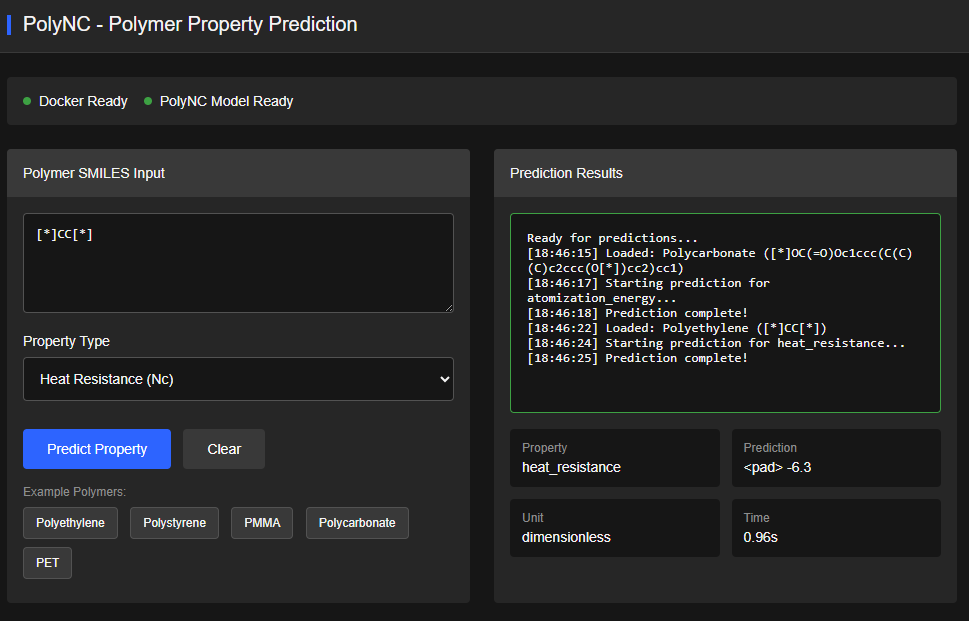
PolyTAO
Apache-2.0 (Free)
A Transformer-Assisted Oriented pretrained model for on-demand polymer generation (conditional generative LLM). Generates polymers with 15 predefined fundamental properties. Achieves ~99.3% chemical validity in top-1 generation.
Features & Details
Qiu, H.; Sun, Z.-Y. On-Demand Reverse Design of Polymers with PolyTAO. npj Computational Materials 10, 273 (2024). DOI:10.1038/s41524-024-01466-5
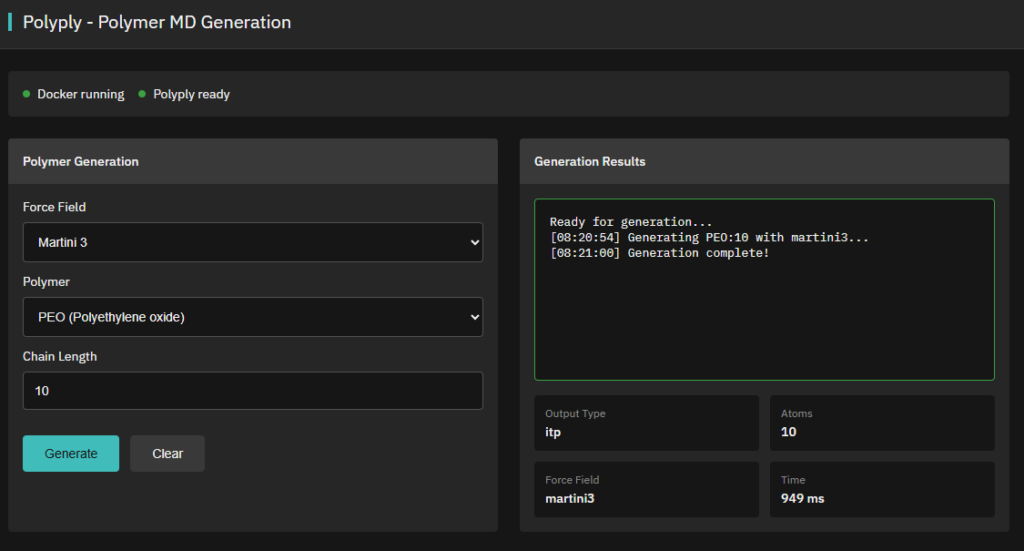
Polyply
Apache-2.0 (Free)
Generates parameters and coordinates for atomistic & coarse-grained polymer MD simulations (force-field and topology agnostic).
Features & Details
Grunewald, F.; Alessandri, R.; Kroon, P.C.; Monticelli, L.; Souza, P.C.T.; Marrink, S.J. Polyply: a python suite for facilitating simulations of (bio-) macromolecules and nanomaterials. Nature Communications 13, 68 (2022). DOI:10.1038/s41467-021-27627-4
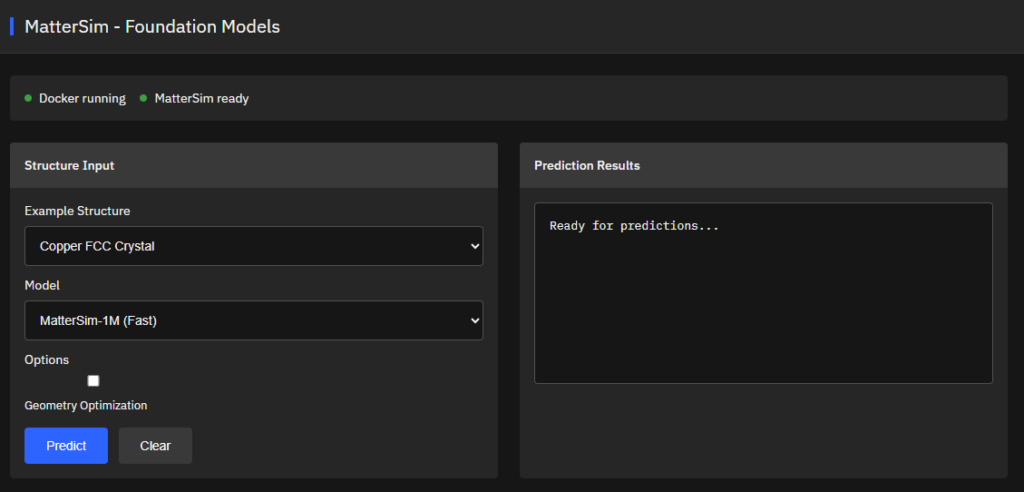
MatterSim
MIT (Free)
Deep learning atomistic foundation model across elements, temperatures, and pressures. Trained on Materials Project and Alexandria datasets with 89 elements (H-Ac) for energy, force, and stress predictions with DFT-level accuracy.
Features & Details
Yang, H. et al. MatterSim: A Deep Learning Atomistic Model Across Elements, Temperatures and Pressures. arXiv:2405.04967 (2024). https://arxiv.org/abs/2405.04967
UI (Human) Applications (5)
UI (Human) Applications
Laboratory operations applications for CMC pharmaceutical development. Electronic lab notebooks, compound databases, equipment inventory, chemical stockroom management, and reactor time series analysis. All apps expose MCP tools for AI-driven automation.
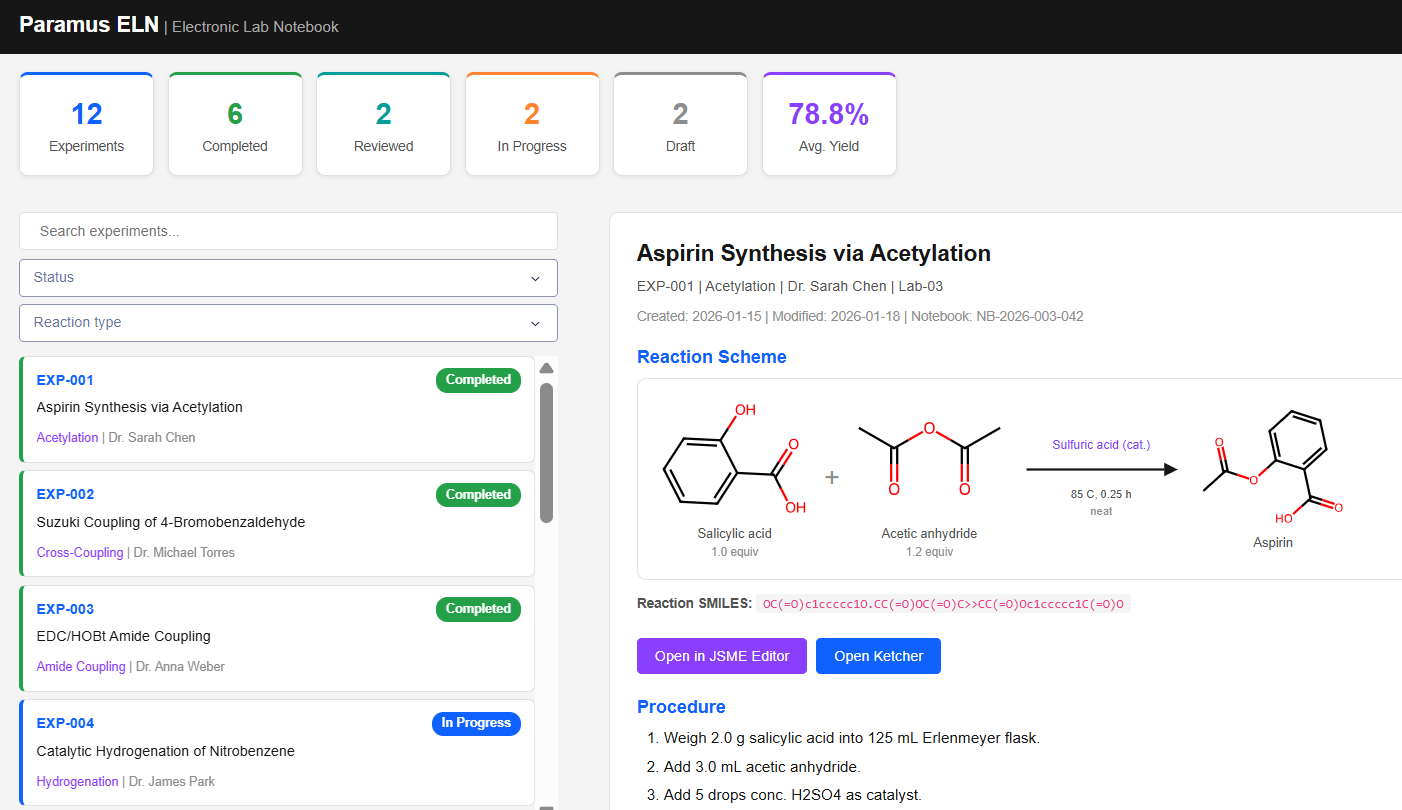
Paramus ELN
MIT (Free)
Electronic Lab Notebook for managing reaction experiments in CMC pharmaceutical development. Includes JSME and Ketcher reaction editors, RDKit-based molecule rendering, and full reaction scheme tracking with SMILES import/export.
Features & Details
Key Features
- Reaction editors: JSME, Ketcher
- RDKit 2D molecule rendering (SVG)
- Status workflow: Draft to Archived
- 12 reaction types: Acetylation, Cross-Coupling, Grignard, Aldol, …
- MCP tools: 6 (list, get, search, statistics, reaction, render)
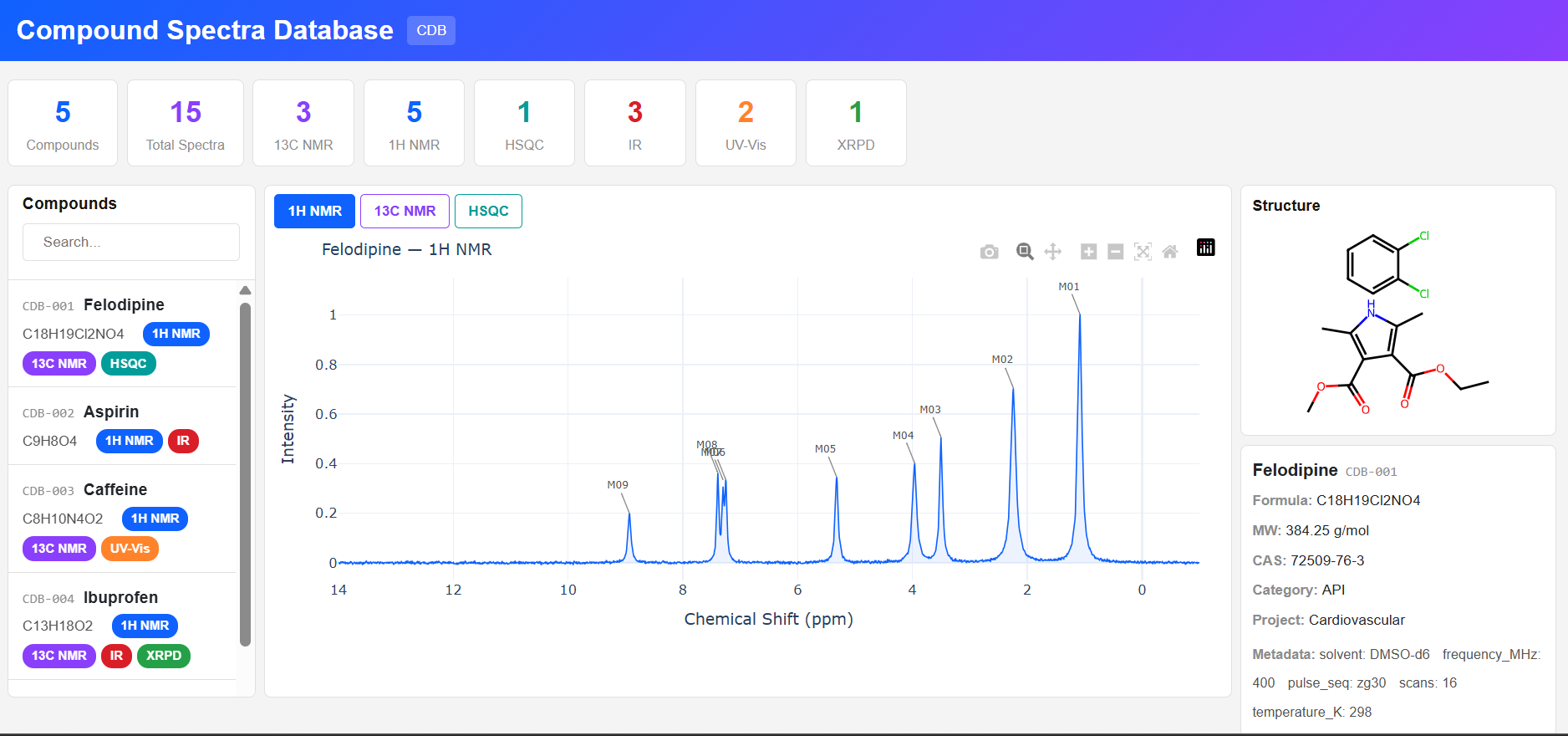
Paramus CDB
MIT (Free)
Compound Spectra Database for browsing compounds and their analytical spectra: NMR (1H, 13C, HSQC, COSY), IR, UV-Vis, XRPD. Interactive Plotly spectrum viewer with realistic Lorentzian/Gaussian line shapes and RDKit structure rendering.
Features & Details
Key Features
- Spectrum types: NMR, IR, UV-Vis, XRPD, generic XY
- Lorentzian/Gaussian spectral data generation
- RDKit 2D structure rendering from SMILES
- Compound search: name, formula, CAS, category
- MCP tools: 4 (list, get, search, statistics)
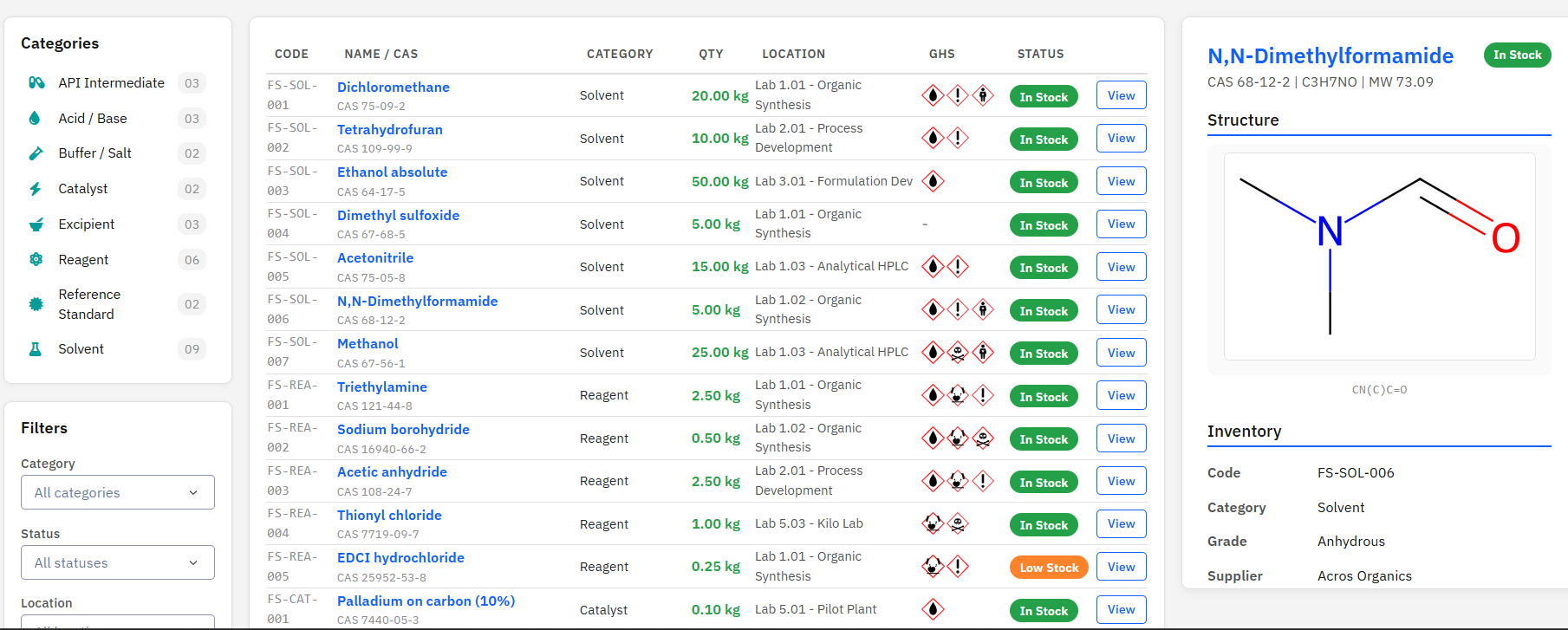
Paramus Stockroom
MIT (Free)
Chemical inventory management for reagents, solvents, catalysts, and raw materials across a 20-lab CMC pharmaceutical work unit. Features GHS hazard pictograms, ADR transport classifications, and RDKit molecule rendering.
Features & Details
Key Features
- 8 chemical categories: Solvent, Reagent, Catalyst, …
- GHS pictograms (GHS01-GHS09) and ADR transport classes
- RDKit 2D structure rendering from SMILES/CAS
- Lot tracking, expiry alerts, supplier management
- MCP tools: 6 (list, get, search, hazard, stats, render)
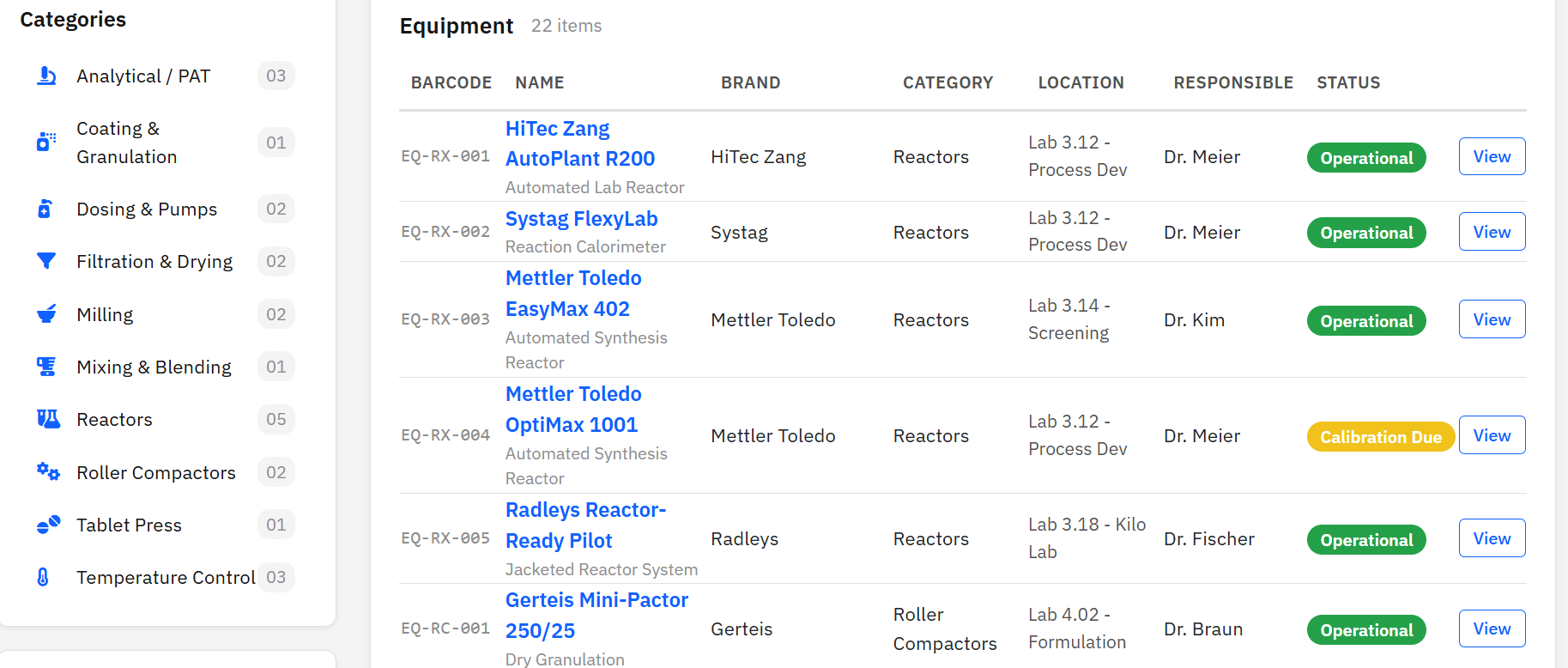
Paramus Inventory
MIT (Free)
Process-scale equipment tracking for CMC pharmaceutical labs. 22 equipment items across 10 categories from manufacturers like HiTec Zang, Systag, and Mettler Toledo. Calibration scheduling, service history, and barcode identification.
Features & Details
Key Features
- 10 categories: Reactors, Filtration, Milling, Analytical, …
- Status: Operational, Maintenance, Calibration Due
- Calibration scheduling and service history
- Barcode identification (EQ-XX-NNN)
- MCP tools: 4 (list, get, search, statistics)
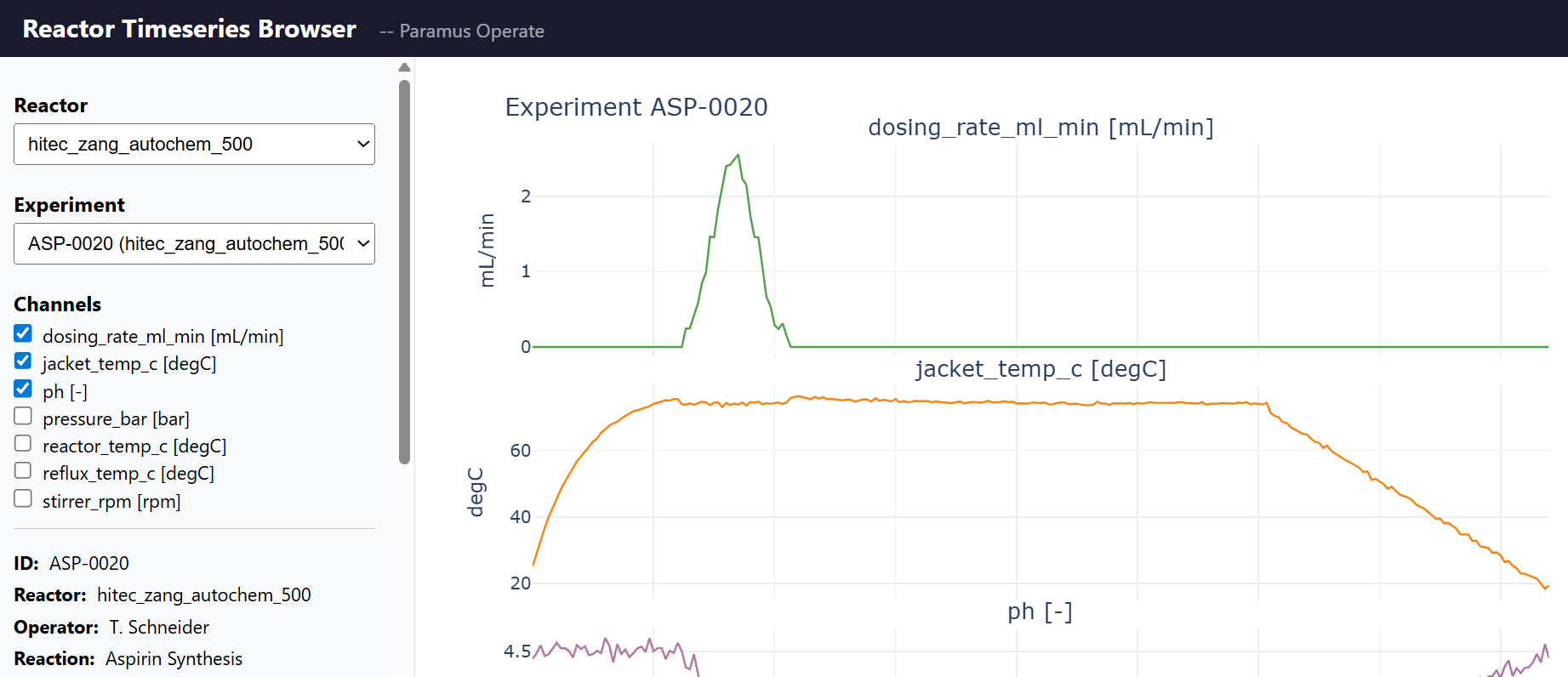
Reactor Timeseries
MIT (Free)
Time series data management for chemical reactor experiments. Supports Systag FlexySys and HiTec Zang reactor systems with 8 measurement channels. Pandas-based data science API for querying, resampling, and comparing reactor runs.
Features & Details
Key Features
- 8 channels: temp, pressure, RPM, pH, heat flow, …
- Pandas API: query, resample, compare, export CSV/Parquet
- SQLite-backed storage with WAL mode
- Yield proxy computation (heat integral, pH, temp stats)
- MCP tools: 9 (list, get, timeseries, channels, stats, compare, yield, health)
Datasets (13)
POLY / Polymer
RadonPy Polymer Dataset (PI1070)
Free (BSD 3)
Contains molecular-simulation data for 1,070 amorphous polymers, including computed physical properties such as density, heat capacity, refractive index, and thermal conductivity under defined conditions.
1.6 MB
RadonPy: Automated Physical Property Calculation using All-atom Classical Molecular Dynamics Simulations for Polymer Informatics npj Computational Materials (2022) DOI:10.1038/s41524-022-00906-4
PI1M
A Benchmark Database for Polymer Informatics
Free (MIT License)
A benchmark database containing approximately 1 million synthetic polymer structures generated using a generative model trained on ~12,000 polymers from PolyInfo. Designed to provide data resources for machine learning research in polymer informatics, covering density, glass transition temperature, melting temperature, and dielectric constants prediction tasks.
108.2 MB
Ruimin Ma, Tengfei Luo PI1M: A Benchmark Database for Polymer Informatics (2020) DOI:10.1021/acs.jcim.0c00726
OMG
Open Macromolecular Genome
Free (GPL)
The Open Macromolecular Genome (OMG) is a comprehensive polymer database designed for generative machine learning and synthetically accessible polymer design. OMG contains nearly 12 million chemically distinct constitutional repeating units (CRUs) generated from 77,281 commercially available monomer reactants using 17 canonical polymerization reactions. The database enables property-driven polymer design by providing synthetic pathways, purchasable reactants, and machine learning-compatible polymer representations.
369.5 MB
Kim, S., Schroeder, C. M., Jackson, N. E. Open Macromolecular Genome: Generative Design of Synthetically Accessible Polymers (2023) DOI:10.1021/acspolymersau.3c00003
VipEA
Vertical Ionization Potentials and Electron Affinities Dataset
Free (MIT License)
Computational dataset of vertical ionization potentials (IP) and electron affinities (EA) for polymer copolymers. Generated using xTB calculations for graph-based molecular property prediction of polymeric materials. Contains data for over 10,000 copolymers with associated quantum chemical properties.
6.5 MB
Matteo Aldeghi, Connor W. Coley A graph representation of molecular ensembles for polymer property prediction (2022) DOI:10.1039/D2SC02839E
OMG-Property-Database
Monomer-level Properties for Synthetically Accessible Polymers
Free (MIT License)
Comprehensive database containing monomer-level chemical and physical properties for approximately 12 million synthetically accessible polymers from the Open Macromolecular Genome. Generated through quantum chemistry calculations integrated with active learning to efficiently probe vast chemical space of synthetically feasible polymers. Includes DFT, TD-DFT calculations, conformer geometries, and ML-based property predictions with uncertainties.
40.9 GB
Seonghwan Kim, Charles M. Schroeder, Nicholas E. Jackson Functional monomer design for synthetically accessible polymers (2025) DOI:10.1039/D4SC08617A
PolyIE
Free (Apache 2.0 License)
Annotations of 146 full-length scholarly articles. Each article is annotated with named entities including compound names, property names, property values, and experimental conditions, along with their complex N-ary relations that capture the intricate relationships between materials, properties, and measurement contexts.
6.2 MB
Cheung, J. J., Zhuang, Y., Li, Y., Shetty, P., Zhao, W., Grampurohit, S., Ramprasad, R. & Zhang, C. PolyIE: A Dataset of Information Extraction from Polymer Material Scientific Literature. arXiv preprint (2023). DOI: 10.48550/arXiv.2311.07715
COMP / Quantum
QM9
Quantum Chemistry Structures and Properties of 134 Kilo Molecules
Free (Creative Commons Attribution 4.0)
QM9 is a comprehensive quantum chemistry dataset containing computed geometric, energetic, electronic, and thermodynamic properties for 130,831 stable small organic molecules made up of C, H, O, N, and F. The dataset provides molecular structures (SMILES, coordinates) and quantum chemical properties calculated using density functional theory for benchmarking molecular property prediction methods and quantum chemistry applications.
82.6 MB
Ramakrishnan, R., Dral, P. O., Rupp, M., von Lilienfeld, O. A. Quantum chemistry structures and properties of 134 kilo molecules (2014) DOI:10.1038/sdata.2014.22
MSR-ACC/TAE25
77k Coupled Cluster Atomization Energies for Broad Chemical Space
Free (CDLA-Permissive-2.0)
Microsoft Research Accurate Chemistry Collection (MSR-ACC) presents MSR-ACC/TAE25, a dataset of 76,879 accurate total atomization energies (TAE) of small molecules with up to 5 non-hydrogen elements up to argon, excluding rare-gas atoms. The atomization energies are computed at the CCSD(T)/CBS level with the W1-F12 thermochemical protocol to provide sub-chemical accuracy within ±1 kcal/mol. The dataset exhaustively covers chemical space by enumerating and sampling chemical graphs, avoiding bias towards particular molecular subspaces.
913.7 MB
Sebastian Ehlert, Jan Hermann, Thijs Vogels, Victor Garcia Satorras, Stephanie Lanius, Marwin Segler, Derk P. Kooi, Kenji Takeda, Chin-Wei Huang, Giulia Luise, Rianne van den Berg, Paola Gori-Giorgi, Amir Karton Accurate Chemistry Collection: Coupled cluster atomization energies for broad chemical space (2025) DOI:10.48550/arXiv.2506.14492
INOR / Material Science
COD
Crystallography Open Database
Free (CC0 1.0)
Open-access collection of crystal structures of organic, inorganic, metal-organic compounds and minerals, excluding biopolymers. Contains over 528,000 crystal structure entries with comprehensive crystallographic data in CIF format, derived from experimental measurements and literature sources. The database serves as a comprehensive resource for crystallographic research, materials science, and structural chemistry applications.
91.8 GB
Saulius Gražulis, Daniel Chateigner, Robert T. Downs, Alexandre F. T. Yokochi, Miguel Quirós, Luca Lutterotti, Elena Manakova, Justas Butkus, Peter Moeck, Armel Le Bail Crystallography Open Database – an open-access collection of crystal structures (2009) DOI:10.1107/S0021889809016690
a-Si-24 synthetic dataset
a-Si-24 (Synthetic Amorphous Silicon Dataset
Free (MIT License)
Synthetic dataset of amorphous silicon structures generated via molecular dynamics simulations using melt-quench trajectories. Each structure represents a final MD snapshot and includes forces and energies labeled by an MTP potential. The dataset covers 3,069 structures (1,317,240 atoms) across various quench rates and densities.
652.0 MB
Signatures of Paracrystallinity in Amorphous Silicon (2024) DOI:10.48550/arXiv.2407.16681
Anionic Solvation Dataset
Solvation Free Energies of Anions: From Curated Reference Data to Predictive Models
Free (Creative Commons Attribution 4.0)
Comprehensive dataset for predicting physicochemical properties of ionizable solutes including 8,241 experimental pKa values across 8 solvents, 5,536 computed gas-phase acidities from DLPNO-CCSD(T) calculations, 6,090 solvation free energies of anions, and 6,088 solvation free energies of neutral compounds computed using COSMO-RS. Includes trained graph neural network models for rapid property prediction as alternative to quantum mechanical approaches.
652.0 MB
Thomas Nevolianis, Jonathan W. Zheng, Simon Müller, Matthias Baumann, Sofja Tshepelevitsh, Ivari Kaljurand, Ivo Leito, Irina Smirnova, William H. Green, Kai Leonhard Solvation free energies of anions: from curated reference data to predictive models (2025) DOI:10.26434/chemrxiv-2025-8bj2t-v2
ANYL / Analytics
QM9S
Dataset (QM9 Spectra
Free (Creative Commons Attribution 4.0)
The QM9S dataset is an enhanced version of the popular QM9 dataset, containing quantum chemical properties and molecular spectra for 130,000 small organic molecules. Built upon the original QM9 dataset, QM9S includes re-optimized molecular geometries at B3LYP/def-TZVP level and comprehensive molecular properties including scalars (energy, NPA charges), vectors (dipole moments), 2nd order tensors (Hessian matrix, polarizability), 3rd order tensors (hyperpolarizability), and complete spectroscopic data (IR, Raman, UV-Vis spectra).
24.1 GB
Zou, Z., et al. QM9S, a comprehensive quantum mechanical dataset of molecular spectra for machine learning (2023) DOI:10.1038/s43588-023-00550-y
ORGN / Organic Chemistry
BigSolDB
2.1: Solubility Values for Organic Compounds in Organic Solvents and Water
Free (Creative Commons Attribution 4.0)
112,465 experimentally measured solubility values of 1,525 organic compounds in 218 solvents reported in 1,687 peer-reviewed articles. Temperature range 243-403 K. Includes mole fraction, molar concentration, and LogS values. Companion density dataset with 218 solvent densities at various temperatures.
19.8 MB
Lev Krasnov, Dmitry Malikov, Marina Kiseleva, Sergei Tatarin, Sergey Sosnin, Stanislav Bezzubov BigSolDB 2.0, dataset of solubility values for organic compounds in different solvents (2025) DOI:10.1038/s41597-025-05559-8
Monetize Without Losing Control
Paramus.ai provides a secure marketplace enabling external vendors to monetize their chemistry applications, AI models, and datasets with full cost carry-over and transparent revenue models. Intellectual property remains fully protected; all packages run within the customer’s local infrastructure.
Publish Your Work
Paramus acts as a distribution and licensing platform for HPC applications, AI models, datasets, and LLM packages. Vendors gain access to a qualified R&D audience across academia and industry without operational overhead.
References (44)
References
- [1] OpenMM: Eastman, P. et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLOS Comput. Biol. 13, e1005659 (2017). DOI:10.1371/journal.pcbi.1005659
- [2] Packmol: Martinez, L. et al. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157-2164 (2009). DOI:10.1002/jcc.21224
- [3] NAMD: Phillips, J.C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153, 044130 (2020). DOI:10.1063/5.0014475
- [4] GROMACS: Abraham, M.J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19-25 (2015). DOI:10.1016/j.softx.2015.06.001
- [5] GAMESS: Schmidt, M.W. et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 14, 1347-1363 (1993). DOI:10.1002/jcc.540141112
- [6] NWChem: Valiev, M. et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 181, 1477-1489 (2010). DOI:10.1016/j.cpc.2010.04.018
- [7] AmberTools: Case, D.A. et al. AmberTools. J. Chem. Inf. Model. 63, 6183-6191 (2023). DOI:10.1021/acs.jcim.3c01153
- [8] CHARMM: Brooks, B.R. et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 30, 1545-1614 (2009). DOI:10.1002/jcc.21287
- [9] CP2K: Kuhne, T.D. et al. CP2K: An electronic structure and molecular dynamics software package. J. Chem. Phys. 152, 194103 (2020). DOI:10.1063/5.0007045
- [10] ORCA: Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 12, e1606 (2022). DOI:10.1002/wcms.1606
- [11] LAMMPS: Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 117, 1-19 (1995). DOI:10.1006/jcph.1995.1039
- [12] PSI4: Smith, D.G.A. et al. Psi4 1.4: Open-Source Software for High-Throughput Quantum Chemistry. J. Chem. Phys. 152, 184108 (2020). DOI:10.1063/5.0006002
- [13] PrexSyn: Luo, S. & Coley, C.W. Synthesizability-Constrained Generative Molecular Design. arXiv:2512.00384 (2024). DOI:10.48550/arXiv.2512.00384
- [14] AiZynthFinder: Genheden, S. et al. AiZynthFinder: a fast, robust and flexible open-source software for retrosynthetic planning. J. Cheminform. 12, 70 (2020). DOI:10.1186/s13321-020-00472-1
- [15] OpenModelica: Fritzson, P. et al. The OpenModelica Integrated Environment for Modeling, Simulation, and Model-Based Development. Modeling, Identification and Control. 41(4), 241-295 (2020). DOI:10.4173/mic.2020.4.1
- [16] Reaktoro: Leal, A.M.M. Reaktoro: An open-source unified framework for modeling chemically reactive systems (2015). https://reaktoro.org
- [17] BOSS: Jorgensen, W.L.; Tirado-Rives, J. Molecular modeling of organic and biomolecular systems using BOSS and MCPRO. J. Comput. Chem. 26, 1689-1700 (2005). DOI:10.1002/jcc.20297
- [18] ASE: Larsen, A.H. et al. The Atomic Simulation Environment – A Python library for working with atoms. J. Phys.: Condens. Matter 29, 273002 (2017). DOI:10.1088/1361-648X/aa680e
- [19] BoFire: BASF Digital Solutions GmbH. BoFire: Bayesian Optimization Framework for Industrial Research and Engineering. https://github.com/experimental-design/bofire
- [20] LigParGen: Dodda, L.S. et al. LigParGen web server: an automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 45, W331-W336 (2017). DOI:10.1093/nar/gkx312
- [21] MDAnalysis: Michaud-Agrawal, N. et al. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 32, 2319-2327 (2011). DOI:10.1002/jcc.21787
- [22] MOPAC: Stewart, J.J.P. Optimization of parameters for semiempirical methods VI. J. Mol. Model. 19, 1-32 (2013). DOI:10.1007/s00894-012-1667-x
- [23] Multiwfn: Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580-592 (2012). DOI:10.1002/jcc.22885
- [24] Open Babel: O’Boyle, N.M. et al. Open Babel: An open chemical toolbox. J. Cheminform. 3, 33 (2011). DOI:10.1186/1758-2946-3-33
- [25] PySCF: Sun, Q. et al. PySCF: the Python-based simulations of chemistry framework. WIREs Comput. Mol. Sci. 8, e1340 (2018). DOI:10.1002/wcms.1340
- [26] RMG: Gao, C.W. et al. Reaction Mechanism Generator: Automatic construction of chemical kinetic mechanisms. Comput. Phys. Commun. 203, 212-225 (2016). DOI:10.1016/j.cpc.2016.02.013
- [27] Soar: Laird, J.E. The Soar Cognitive Architecture. MIT Press (2012). ISBN:978-0262122962
- [28] Tinker: Rackers, J.A. et al. Tinker 8: Software Tools for Molecular Design. J. Chem. Theory Comput. 14, 5273-5289 (2018). DOI:10.1021/acs.jctc.8b00529
- [29] xTB: Bannwarth, C. et al. GFN2-xTB – An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method. J. Chem. Theory Comput. 15, 1652-1671 (2019). DOI:10.1021/acs.jctc.8b01176
- [30] OpenFOAM: Weller, H.G. et al. A tensorial approach to computational continuum mechanics using object-oriented techniques. Computers in Physics. 12, 620-631 (1998). DOI:10.1063/1.168744
- [31] OpenQBMM: Passalacqua, A. et al. An open-source quadrature-based population balance solver for OpenFOAM. Chemical Engineering Science. 176, 306-318 (2018). DOI:10.1016/j.ces.2017.10.043
- [32] DWSIM: Wagner, D. “DWSIM – An Open-Source Chemical Process Simulator” https://dwsim.org/
- [33] Code_Saturne: Archambeau, F. et al. Code_Saturne: A Finite Volume Code for the computation of turbulent incompressible flows. International Journal on Finite Volumes. 1(1), 1-62 (2004).
- [34] Ether0:q2_k: Narayanan, S.M. et al. Training a Scientific Reasoning Model for Chemistry. arXiv:2506.17238 (2025). DOI:10.48550/arXiv.2506.17238
- [35] Llama 3.2:1b: Meta AI. Llama 3.2 1B (1.23-billion-parameter multilingual language model), released 25 September 2024, https://huggingface.co/meta-llama/Llama-3.2-1B
- [36] DeepSeek R1:8b: DeepSeek-AI, Guo, D., Yang, D., Zhang, H., et al. DeepSeek-R1: Incentivizing Reasoning Capability in LLMs via Reinforcement Learning. arXiv:2501.12948 (2025). DOI:10.48550/arXiv.2501.12948
- [37] AIMNet2: Anstine, D.M.; Isayev, O. AIMNet2: A Neural Network Potential to Meet your Neutral, Charged, Organic, and Elemental-Organic Needs. J. Phys. Chem. A (2023). DOI:10.1021/acs.jpca.2c06685
- [38] MACE: Batatia, I. et al. MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields. NeurIPS (2022). DOI:10.48550/arXiv.2206.07697
- [39] ORB: Rhodes, B. et al. Orb-v3: atomistic simulation at scale. arXiv:2504.06231 (2025).
- [40] TransPolymer: Xu, C.; Wang, Y.; Barati Farimani, A. TransPolymer: a Transformer-based language model for polymer property predictions. npj Computational Materials 9, 64 (2023). DOI:10.1038/s41524-023-01009-9
- [41] PolyNC: Qiu, H.; Liu, L.; Qiu, X.; Dai, X.; Ji, X.; Sun, Z.-Y. PolyNC: a natural and chemical language model for unified polymer properties prediction. Chemical Science (2024). DOI:10.1039/D3SC05079C
- [42] PolyTAO: Qiu, H.; Sun, Z.-Y. On-Demand Reverse Design of Polymers with PolyTAO. npj Computational Materials 10, 273 (2024). DOI:10.1038/s41524-024-01466-5
- [43] Polyply: Grunewald, F.; Alessandri, R.; Kroon, P.C.; Monticelli, L.; Souza, P.C.T.; Marrink, S.J. Polyply: a python suite for facilitating simulations of (bio-) macromolecules and nanomaterials. Nature Communications 13, 68 (2022). DOI:10.1038/s41467-021-27627-4
- [44] MatterSim: Yang, H. et al. MatterSim: A Deep Learning Atomistic Model Across Elements, Temperatures and Pressures. arXiv:2405.04967 (2024). https://arxiv.org/abs/2405.04967