Model Marketplace
The Paramus AI Marketplace provides a curated portfolio of ‘classic’ chemistry AI models, HPC (computing) applications, and local LLMs. All this enables scientific teams to integrate advanced calculations and AI into large-scale chemistry workflows. They all run ‘out of the box’. Also the agents an mount/pull/install/uninstall the appliacations.
Computing Applications (HPC)
HPC enables scalable simulation, modeling, and analysis of chemical systems. In quantum chemistry (QC), HPC is crucial for performing accurate electronic structure calculations at high theory levels, enabling reliable predictions for molecular design and reactivity.

OpenMM
MIT (Free)
A molecular dynamics toolkit for simulating biomolecules, providing energy minimization, NVT/NPT ensemble sampling, and support for AMBER and CHARMM force fields with GPU-accelerated calculations via CUDA and OpenCL.
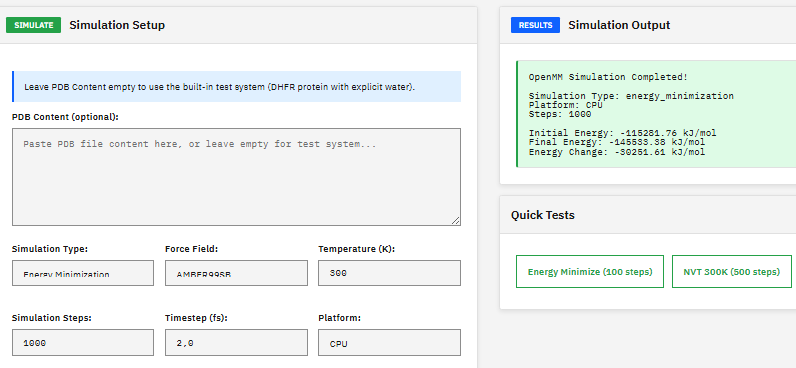
Eastman, P. et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLOS Comput. Biol. 13, e1005659 (2017). DOI:10.1371/journal.pcbi.1005659
Packmol
MIT (Free)
A molecular packing tool that creates initial configurations for molecular dynamics simulations by positioning molecules into defined geometric regions (boxes, spheres, cylinders) while avoiding atomic overlaps and steric clashes.
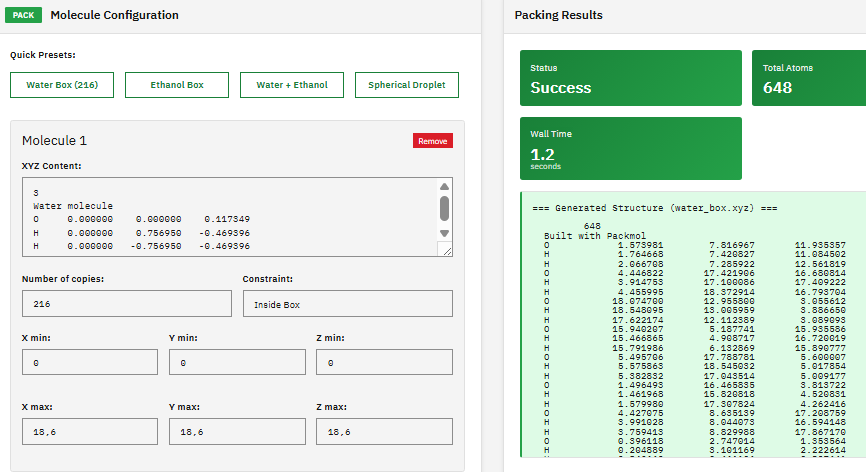
Martinez, L. et al. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157-2164 (2009). DOI:10.1002/jcc.21224
NAMD
UIUC-NAMD (free academic, commercial license required)
A parallel molecular dynamics engine for simulating large biomolecular systems, supporting CHARMM, AMBER, and OPLS force fields with free energy perturbation, replica exchange, and QM/MM.
Phillips, J.C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153, 044130 (2020). DOI:10.1063/5.0014475
License → https://www.ks.uiuc.edu/Research/namd
GROMACS
LGPL-2.1 (Free)
A molecular dynamics package for simulating proteins, lipids, and nucleic acids, with multi-level parallelism supporting CUDA GPU acceleration, MPI distributed computing, and OpenMP threading.
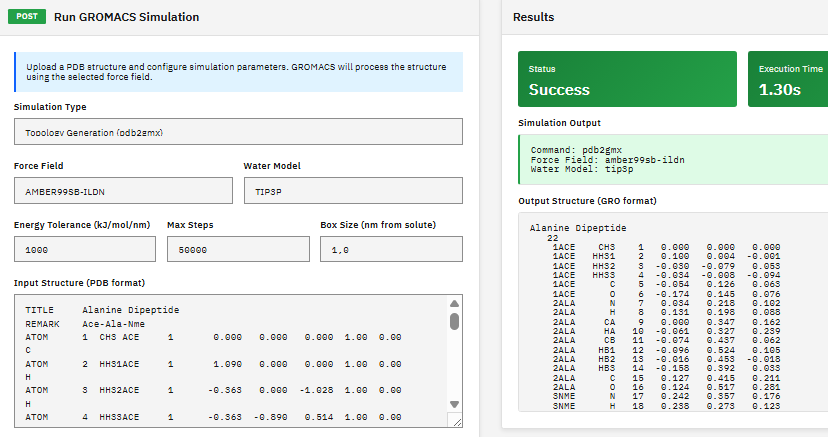
Abraham, M.J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19-25 (2015). DOI:10.1016/j.softx.2015.06.001
GAMESS
Proprietary Academic License
An ab initio quantum chemistry package (General Atomic and Molecular Electronic Structure System) supporting Hartree-Fock, DFT, MP2, coupled cluster, MCSCF, and CI methods for electronic structure calculations, geometry optimizations, and property predictions.
Schmidt, M.W. et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 14, 1347-1363 (1993). DOI:10.1002/jcc.540141112
Registration required → https://www.msg.chem.iastate.edu/GAMESS
NWChem
ECL-2.0 (Free)
A computational chemistry platform providing Hartree-Fock, DFT, MP2, and coupled cluster methods with MPI parallelization for electronic structure simulations on computing clusters.
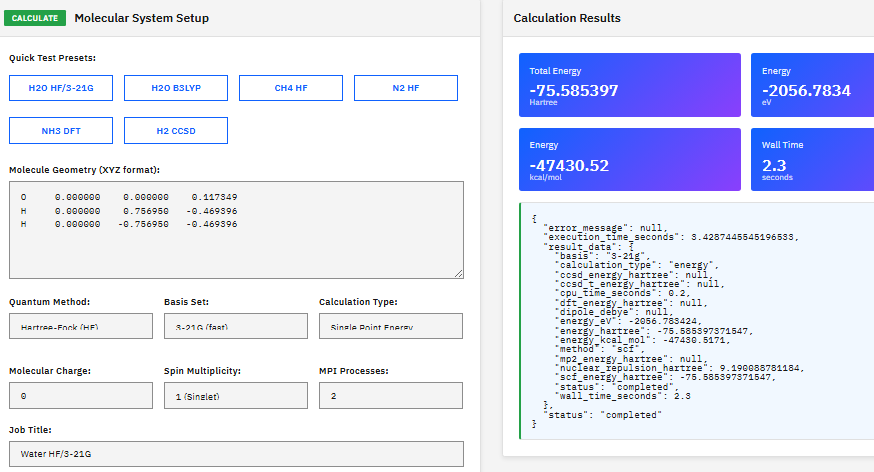
Valiev, M. et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 181, 1477-1489 (2010).
DOI:10.1016/j.cpc.2010.04.018
AmberTools
GPL-2.0 (Free)
A force field parameterization and molecular dynamics preparation suite providing antechamber for GAFF/GAFF2 atom typing, tleap for building simulation systems, cpptraj for trajectory analysis, and reduce for hydrogen optimization.
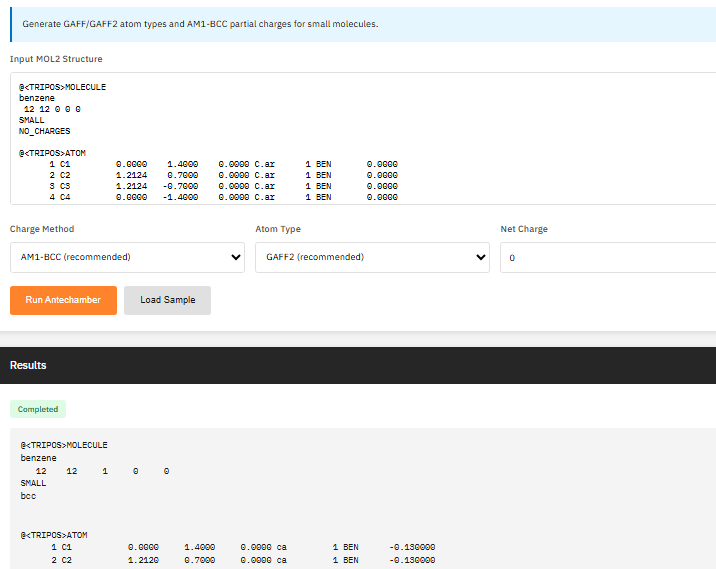
Case, D.A. et al. AmberTools. J. Chem. Inf. Model. 63, 6183-6191 (2023). DOI:10.1021/acs.jcim.3c01153
CHARMM
Academic/Non-profit
A molecular simulation program (Chemistry at HARvard Macromolecular Mechanics) for modeling macromolecular systems, providing energy calculations, minimization, dynamics simulations, and sampling methods for proteins, nucleic acids, lipids, and small molecules.
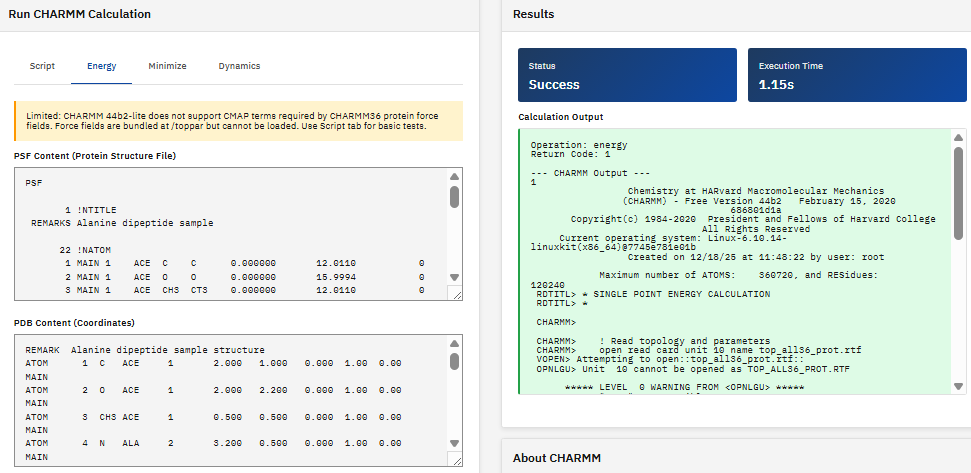
Brooks, B.R. et al. CHARMM: The Biomolecular Simulation Program. J. Comput. Chem. 30, 1545-1614 (2009). DOI:10.1002/jcc.21287
Registration required → https://academiccharmm.org
CP2K
GPL-2.0 (Free)
A quantum chemistry and solid-state physics package combining DFT with Gaussian and plane-wave (GPW/GAPW) methods, enabling geometry optimization, cell optimization, and ab initio molecular dynamics for both periodic crystals and isolated molecular systems.
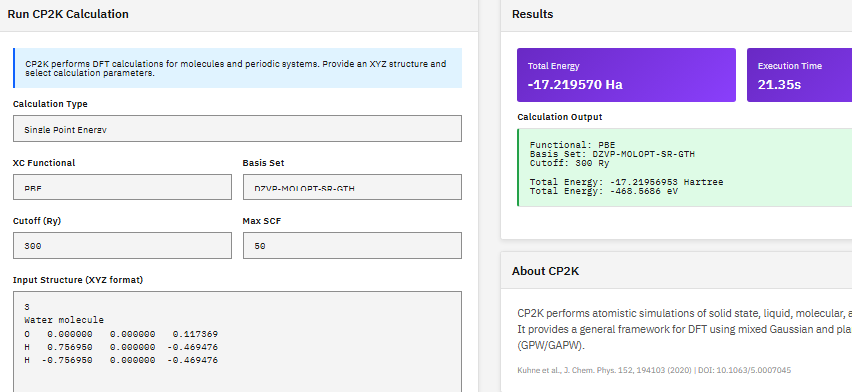
Kuhne, T.D. et al. CP2K: An electronic structure and molecular dynamics software package. J. Chem. Phys. 152, 194103 (2020). DOI:10.1063/5.0007045
ORCA
Free academic, commercial license required
A versatile quantum chemistry program supporting DFT, ab initio, and semi-empirical methods. ORCA enables accurate calculations of molecular structures, spectra, and reaction mechanisms and serves as a backend for AI-assisted computational chemistry workflows in PARAMUS.
Neese, F.
The ORCA program system. WIREs Comput. Mol. Sci. 12, e1606 (2022).
DOI:10.1002/wcms.1606
License → https://orcaforum.kofo.mpg.de
LAMMPS
Academic, non-commercial use only
A molecular simulation engine (LAMMPS-oriented) that encodes atomistic polymer systems into descriptors (“fingerprints”) enabling fast property predictions, structure–property correlations, and integration into polymer informatics workflows.
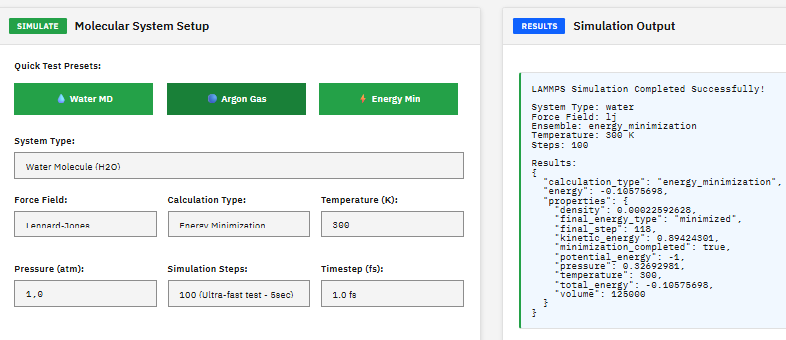
C.; Plimpton, S. LAMMPS: A molecular dynamics engine for scalable simulations of materials and polymers. J. Comput. Phys. 117, 1–19 (1995). DOI:10.1006/jcph.1995.1039 / arXiv:0808.2505
PSI4
LGPL-3.0 (Free)
A quantum chemistry suite providing Hartree-Fock, DFT, MP2, coupled cluster, and symmetry-adapted perturbation theory (SAPT) methods for high-throughput electronic structure calculations.
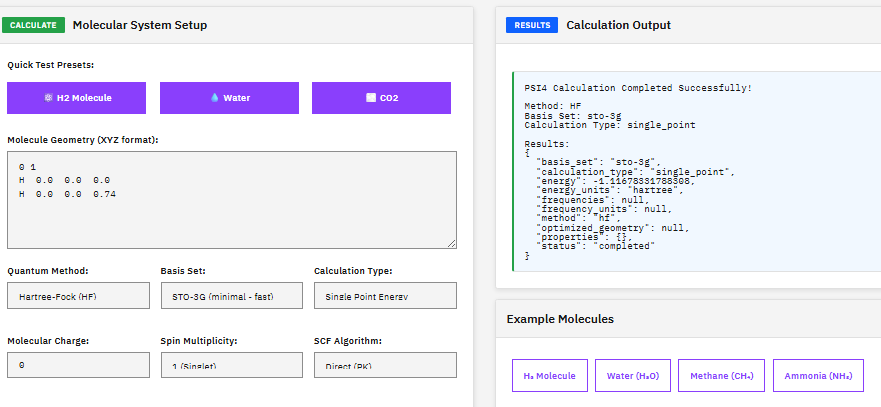
D. G. A. Smith et al., “Psi4 1.4: Open-Source Software for High-Throughput Quantum Chemistry,” J. Chem. Phys. 152, 184108 (2020)
AiZynth Finder
MIT (Free)
AI-driven retrosynthetic route planning tool for computer-aided synthesis using Monte Carlo tree search and deep neural networks to predict multi-step synthesis routes for organic molecules with route scoring and ranking capabilities.
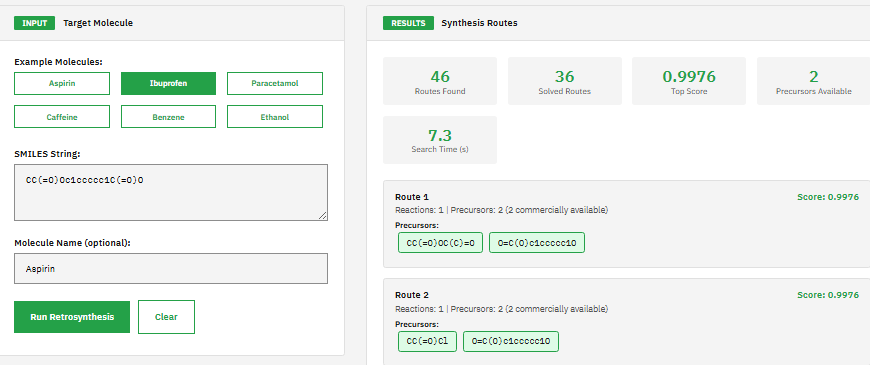
Genheden, S. et al. AiZynthFinder: a fast, robust and flexible open-source software for retrosynthetic planning. J. Cheminform. 12, 70 (2020).
DOI:10.1186/s13321-020-00472-1
Languange Models (LLM)
Running large language models locally is a statement of independence: data stays private, inference remains under full control, and chemistry does not leave the laboratory.
The price is: speed. Today it is painfully slow. But it works.
Ether0:q2_k
Apache-2.0 (Free)
Scientific research language model fine-tuned for chemistry, biology, and materials science. Based on Qwen2.5-24B with Q2_K quantization optimized for single-CPU execution, enabling research workflows on standard hardware.
8.9GB, optimized for single-CPU systems
Narayanan, S.M. et al. Training a Scientific Reasoning Model for Chemistry. arXiv:2506.17238 (2025). DOI:10.48550/arXiv.2506.17238
Llama 3.2:1b
Llama 3.2 Community License
Commercial use with specified attribution
Provides lightweight natural language understanding and generation suitable for embedded and edge devices. Its compact size enables efficient inference on consumer-grade GPUs and even high-performance CPUs. Despite its small footprint, it supports multilingual text processing, basic reasoning, and customizable fine-tuning for specialized tasks.
Meta AI. Llama 3.2 1B (1.23-billion-parameter multilingual language model), released 25 September 2024, https://huggingface.co/meta-llama/Llama-3.2-1B
DeepSeek R1:8b
MIT (Free)
A reasoning-focused large language model for scientific and technical tasks, offering chain-of-thought reasoning and step-by-step problem decomposition for general questions.
DeepSeek-AI, Guo, D., Yang, D., Zhang, H., et al. DeepSeek-R1: Incentivizing Reasoning Capability in LLMs via Reinforcement Learning. arXiv:2501.12948 (2025). DOI:10.48550/arXiv.2501.12948
AI Models
This models are ‘one-click installable programs’ following the Paramus AI Runtime Specification.
AIMNet2
Free (MIT)
High-accuracy neural network potential for organic and elemental-organic molecules supporting neutral and charged species. Predicts energies, forces, and partial charges at wB97M-D3BJ/def2-TZVPP level for drug discovery and computational chemistry applications.
367 MB
Anstine, D.M.; Isayev, O. AIMNet2: A Neural Network Potential to Meet your Neutral, Charged, Organic, and Elemental-Organic Needs. J. Phys. Chem. A (2023).
DOI:10.1021/acs.jpca.2c06685
MACE*)
Free (Apache 2.0)
Equivariant message passing neural networks achieving state-of-the-art accuracy for atomistic simulations. Universal foundation models for molecular dynamics and materials science with DFT-level accuracy and computational efficiency.
936 MB
Batatia, I. et al. MACE: Higher Order Equivariant Message Passing Neural Networks for Fast and Accurate Force Fields. NeurIPS 2022. DOI:10.48550/arXiv.2206.07697
ORB*)
Apache-2.0 (Free)
Neural network potentials for atomic simulations providing fast and accurate energy/force predictions for materials science, chemistry, and molecular systems. Force fields supporting structure optimization, molecular dynamics, and property calculations.
1.9 GB ![]()
Rhodes, B. et al. Orb-v3: atomistic simulation at scale. arXiv:2504.06231 (2025).
TransPolymer
Free (MIT)
Predicts polymer properties using transformer-based deep learning models trained on polymer structure–property datasets. Designed for inverse design and polymer informatics workflows.
562 MB
Xu, C.; Wang, Y.; Barati Farimani, A.
“TransPolymer: a Transformer-based language model for polymer property predictions.” npj Computational Materials 9, 64 (2023).
DOI:10.1038/s41524-023-01009-9
PolyNC
Free (Apache 2.0)
A unified natural & chemical language model (text-to-text) for predicting polymer properties (multi-task: regression + classification).
1.02 GB ![]()
Qiu, H.; Liu, L.; Qiu, X.; Dai, X.; Ji, X.; Sun, Z.-Y.
“PolyNC: a natural and chemical language model for unified polymer properties prediction.” Chemical Science (2024).
DOI: 10.1039/D3SC05079C
PolyTAO
Free (Apache 2.0)
A Transformer-Assisted Oriented pretrained model for on-demand polymer generation (conditional generative LLM). Generates polymers with 15 predefined fundamental properties. Achieves ~99.3 % chemical validity in top-1 generation.
Qiu, H.; Sun, Z.-Y.
“On-Demand Reverse Design of Polymers with PolyTAO.” npj Computational Materials 10, 273 (2024). DOI:10.1038/s41524-024-01466-5
Polyply
Free (Apache 2.0)
Generates parameters and coordinates for atomistic & coarse-grained polymer MD simulations (force-field and topology agnostic)
Grünewald, F.; Alessandri, R.; Kroon, P. C.; Monticelli, L.; Souza, P. C. T.; Marrink, S. J. “Polyply: a python suite for facilitating simulations of (bio-) macromolecules and nanomaterials.” Nature Communications 13, 68 (2022). DOI:10.1038/s41467-021-27627-4
MatterSim*)
Free (MIT)
Deep learning atomistic foundation model across elements, temperatures, and pressures. Trained on Materials Project and Alexandria datasets with 89 elements (H-Ac) for energy, force, and stress predictions with DFT-level accuracy.
Yang, H. et al. MatterSim: A Deep Learning Atomistic Model Across Elements, Temperatures and Pressures. arXiv:2405.04967 (2024).
https://arxiv.org/abs/2405.04967
Reference Model
Free
This model implements a reference for the Paramus light AI Model specification 1.2 Used for system performance (telemetry)
Model Variants
Some of the AI modles have varaints. PARAMUS automatically selects the best model because the ‘Proven Acceptable Model Ranges’ are considered. Also telemetry data (system performance) is taken into account for optimal selection.
*) MACE Model Variants
- MACE-MP-0: 89 elements, Materials Project, PBE+U/VASP
- MACE-OFF-23: 10 elements, organic molecules, SPICE dataset, ORCA
- MACE-MPA-0: 118 elements, multi-fidelity materials
- MACE-OMAT-0: 118 elements, OpenMat corpus
- MACE-MatPES-PBE-0: 89 elements, PBE+D3/VASP
- MACE-MatPES-r2SCAN-0: 89 elements, r2SCAN meta-GGA/VASP
*) MatterSim Model Variants
- MatterSim-1M: 1M parameters, faster inference, lower memory
- MatterSim-5M: 5M parameters, higher accuracy
*) ORB OMol Variants (organic molecules and small molecular systems)
- orb-v3-conservative-omol: Backprop forces, higher accuracy for MD
- orb-v3-direct-omol: Direct forces, 4x faster inference, lower memory
*) ORB OMAT Variants (inorganic materials and crystals)
- orb-v3-conservative-inf-omat: Backprop forces, unlimited neighbors, max accuracy
- orb-v3-conservative-20-omat: Backprop forces, 20Å cutoff, faster
- orb-v3-direct-inf-omat: Direct forces, unlimited neighbors, 4x faster, lower memory
- orb-v3-direct-20-omat: Direct forces, 20Å cutoff, fastest
*) ORB MPA Variants [broader chemistry (MPTraj + Alexandria datasets)]
- orb-v3-conservative-inf-mpa: Backprop forces, unlimited neighbors, max accuracy
- orb-v3-conservative-20-mpa: Backprop forces, 20Å cutoff, faster
- orb-v3-direct-inf-mpa: Direct forces, unlimited neighbors, 4x faster, lower memory
- orb-v3-direct-20-mpa: Direct forces, 20Å cutoff, fastest
Monetize Without Losing Control
Paramus.ai provides a secure AI model marketplace enabling external vendors to monetize their models with full cost carry-over and transparent revenue models. Intellectual property remains fully protected;
Empower Your Models
Paramus acts only as a distribution and licensing platform. Vendors gain access to a qualified R&D audience across academia and industry without operational overhead.