Computational Chemistry Agent
The Computational Chemistry Agent is a digital assistant designed for performing quantum chemical calculations. It executes molecular dynamics simulations, predicts reaction kinetics and thermodynamics, and computes theoretical spectra. The tool provides a streamlined interface for automating complex computational chemistry tasks.
The system integrates several open-source libraries, widely used quantum chemistry software packages. PSI4 is an open-source platform. The agent leverages these tools to deliver high-accuracy quantum mechanical calculations.
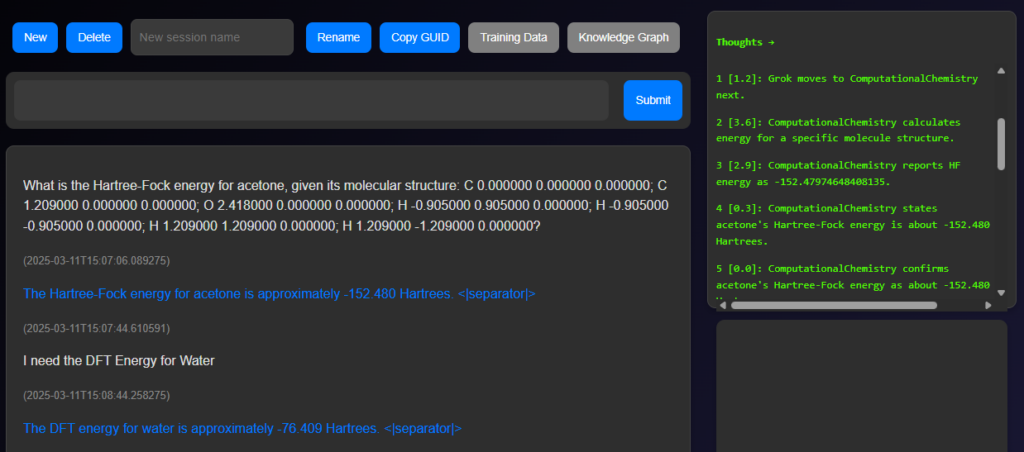
PARAMUS calling Computational Chemistry Calculations
This agent is currently in an experimental phase, meaning stability and performance may vary. Users should be aware of potential limitations and ensure compatibility with their specific computational needs. Future updates aim to enhance robustness and extend functionality.
Remote Batch Service
The Computational Chemistry Agent utilizes a remote batch service for executing PSI4 calculations on a dedicated high-performance computing system. Due to the computational demands of quantum chemistry, these tasks are offloaded to a remote server with optimized hardware for large-scale simulations. This infrastructure ensures fast and accurate processing of complex molecular computations.
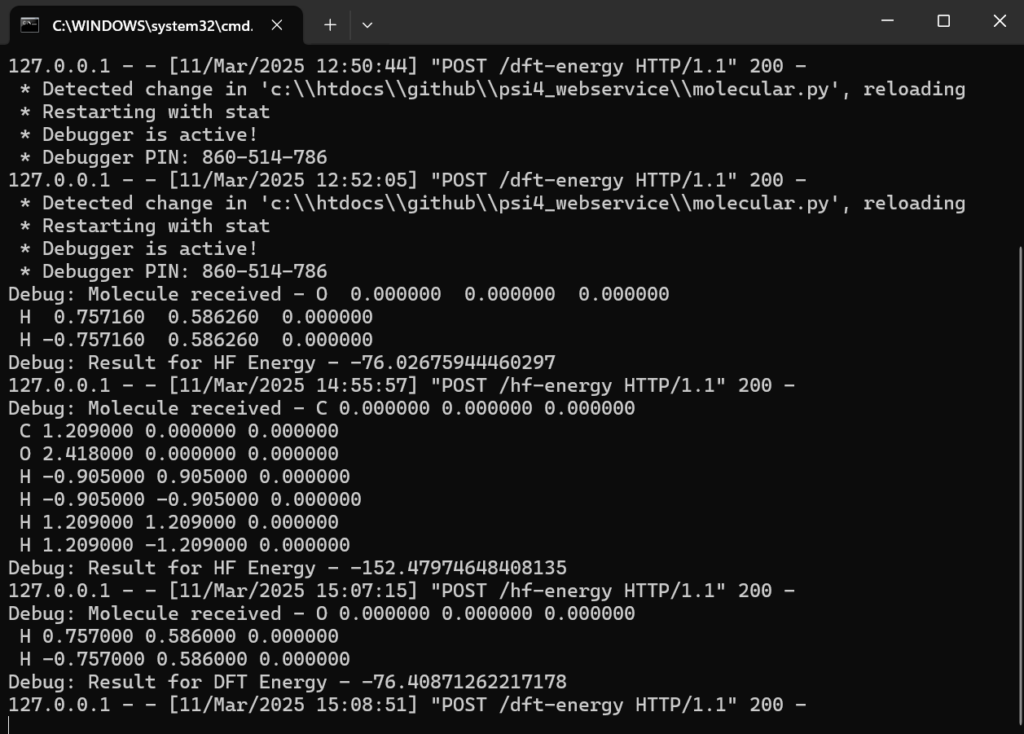
PSI4 Service on remote computer
By leveraging this remote execution model, the agent provides access to PSI4 without requiring local high-performance computing resources. This approach allows researchers and professionals to perform advanced calculations without investing in expensive local hardware.